Young Individual Research Grant (YIRG) Awardees

We use integrative molecular modelling and molecular dynamics simulation to learn how large biological macromolecules such as antibodies and protein complexes behave in their native environment, which can subsequently be exploited for therapeutic interventions.
Monoclonal antibodies have been an instrumental part of standard chemotherapy regiments. Apart from cancer therapeutics, antibodies also show great potential in diagnosis due to their remarkable specificity. To date, most antibodies used in cancer therapy and diagnosis belong to the immunoglobulin (Ig) isotype IgG. The use of other antibody isotypes including IgM remains largely unexplored despite their potential benefits. Unlike IgG, which has two antigen-binding fragment (Fab) regions, IgM has up to twelve Fabs and thus, in principle, a higher binding avidity to its antigen. Due to its large size, IgM can also form bridges between distant antigens on different cancer cells to trigger agglutination, which is beneficial to neutralise metastatic cells, and for enrichment of circulating tumour cells in blood samples for diagnosis. IgM is therefore an attractive candidate for future cancer treatment and diagnostic tools.
Our collaborator from APD lab in EDDC showed that the IgM form of Pertuzumab and Trastuzumab, monoclonal antibodies used to treat breast cancer, have different binding avidities to their target, human epidermal growth factor receptor 2 (HER2). We built integrative models of full-length Pertuzumab and Trastuzumab IgM hexamers bound to the HER2 extracellular domains (ECD). Our models revealed that the Pertuzumab IgM hexamer can utilize all its Fab domains to bind to twelve HER2 ECDs simultaneously. The binding was shown to be stable in subsequent simulations. Conversely, we found that the Trastuzumab IgM hexamer was not able to bind multiple HER2 ECDs due to steric clashes caused by the large globular domain of the latter (Figure 1). This computational prediction was then validated by cell count assays using HER2 over expressing cell lines showing that Pertuzumab IgM was better than IgG at inhibiting cell proliferation, whereas the opposite trend prevailed for Trastuzumab.
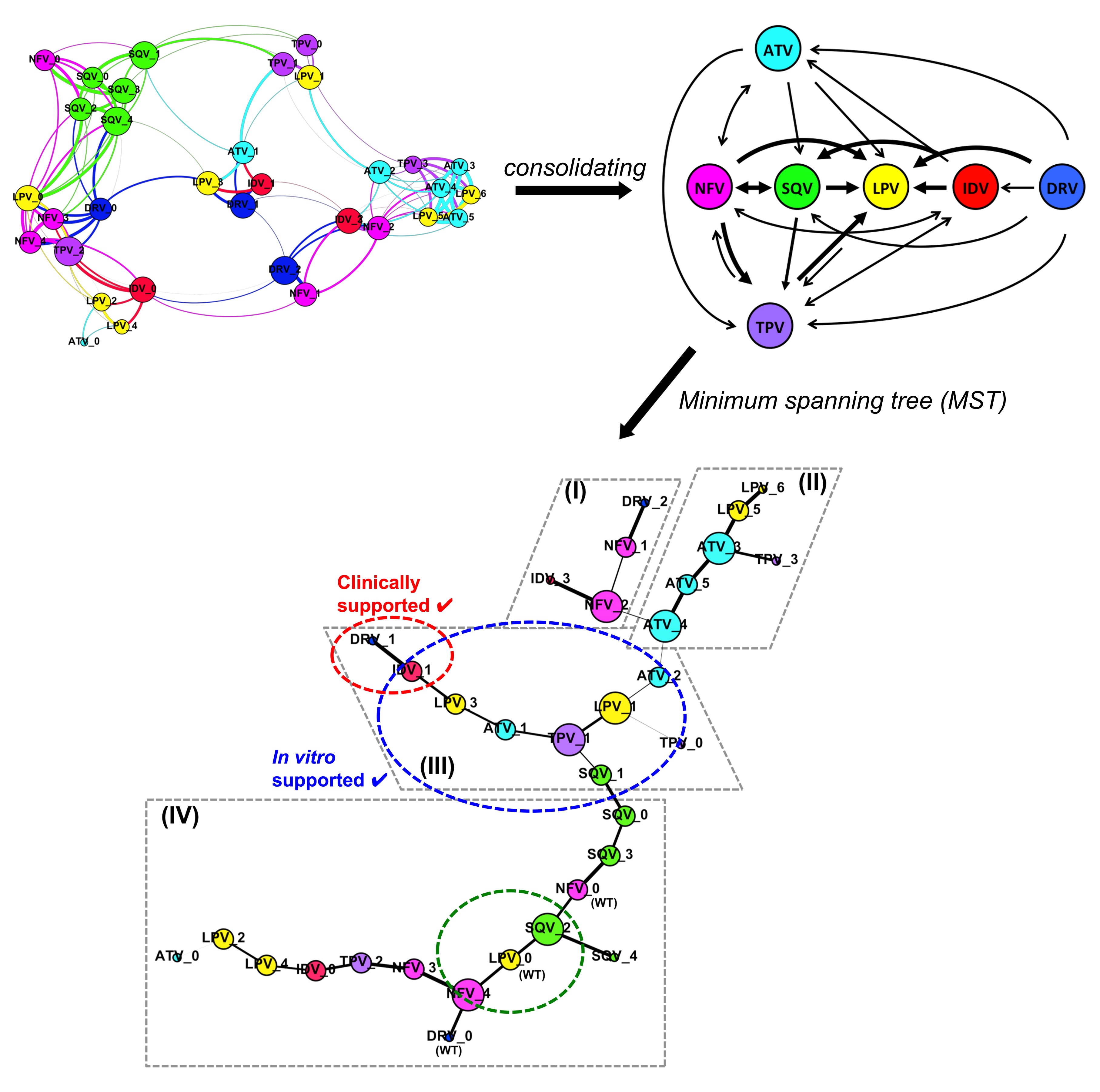
Based on the structural responses and viral fitness cost of the clinically reported HIV-1 protease mutations, we reported a PI resistance-related pathway that the protease might undertake to cross-resist among current PIs in the anti-retroviral therapy.
We found the structural rationale for the rapid development of cross-resistance among the seven clinically used PIs, e.g. Atazanavir (ATV), Darunavir (DRV), Indinavir (IDV), Lopinavir (LPV), Nelfinavir (NFV), Saquinavir (SQV), and Tipranavir (TPV). Among these, some PIs would be better used in clinical settings against naïve HIV-1 infections or when resistance has already been developed towards another PI.
Our findings suggest the use of LPV as the first line of PI in the current anti-retroviral therapy. In circumstances with the emergence of PI-resistant mutations, certain other drugs would be more useful in subsequent lines of treatment (Figure 1). Hence the study provides a structural understanding that may be useful to guide the clinical PI use in the therapy, aiding in drug selection to prolong the effectiveness of the given PI.
Reviewing HIV-1 Gag Mutations in Protease Inhibitors Resistance
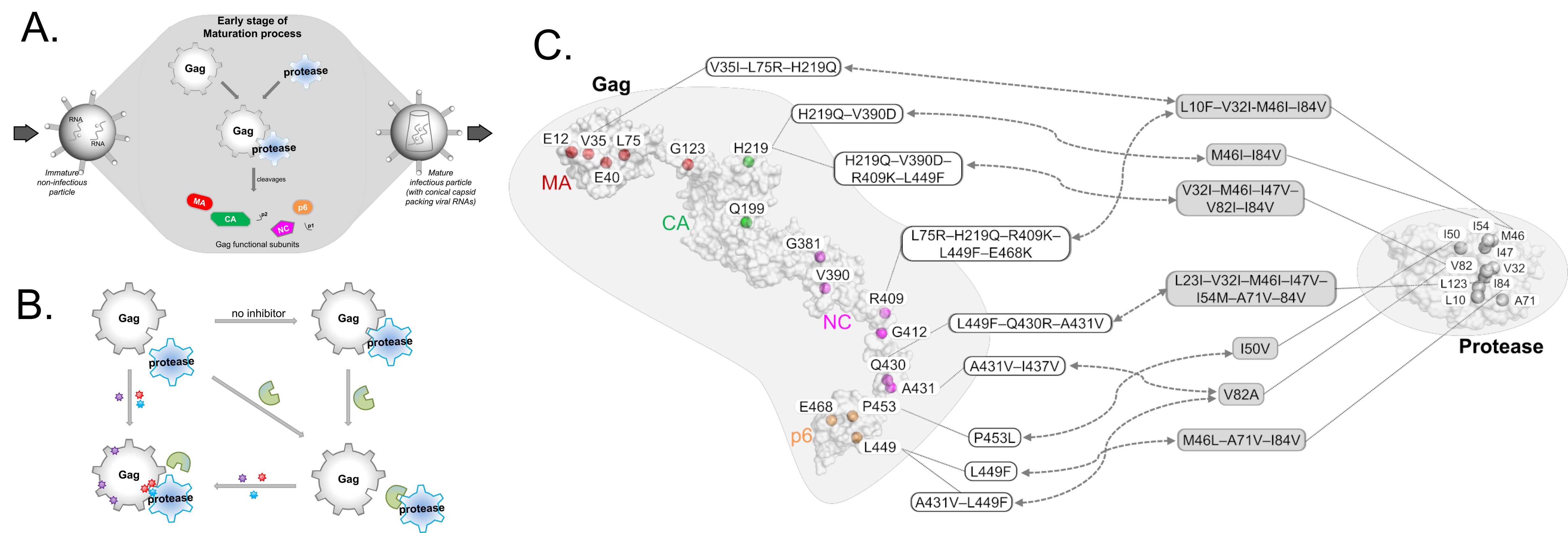
HIV-1 protease inhibitors against the viral protease are often hampered by drug resistance mutations in protease and in the protease substrate Gag (Figure 2A-B). To overcome this drug resistance, it may be more efficient to target Gag alongside with protease rather than targeting protease alone. To inhibit Gag, understanding of its drug resistance mutations and the corresponding structural changes on protease binding thus needs investigating.
While mutations on Gag have already been mapped to protease inhibitor resistance (Figure 2C), there remain many mutations, particularly those outside the Gag cleavage sites, that are not well-characterized. Through structural studies to unravel how Gag mutations contributes to protease drug resistance synergistically, it is thus possible to glean insights to design novel Gag inhibitors. In this review (Su et al., 2019), we discuss the structural role of both novel and previously reported Gag mutations in PI resistance, and how new Gag inhibitors can be designed.
We employ computational methods to understand molecular details of macromolecular assemblies such as viruses and virus like particles. Our computational predictions aim at explaining often low resolution experimental observations in order to understand various biological phenomena. Using in silico modelling, our objective is to stimulate new experiments which together deliver new biological insights.
Dengue virus (DENV), a member of the flavivirus genus, infects hundreds of millions of people worldwide every year. The outermost layer of a flavivirus is made up of envelope (E) proteins embedded in a lipid membrane, while the inner core contains capsid proteins in complex with an RNA genome. E proteins are crucial in viral infection since through them the virus attaches to the host cell. They are also engaged in antibody binding which can help to prevent infection. At present, there are no approved drugs or highly effective and safe vaccines for DENV. Virus like particles (VLPs) represent novel promising platforms as vaccine alternatives because they are non-infectious and highly immunogenic. Using the first ever DENV VLP experimental structure, we are employing a computational approach to redesign VLPs with new epitopes for enhanced therapeutic stability and efficiency. Our computational predictions of new mutations on DENV VLPs are being experimentally engineered and tested at the Graduate Institute of Veterinary Public Health, National Chung Hsing University, Taiwan.
FLAVIVIRUS LIFE CYCLE
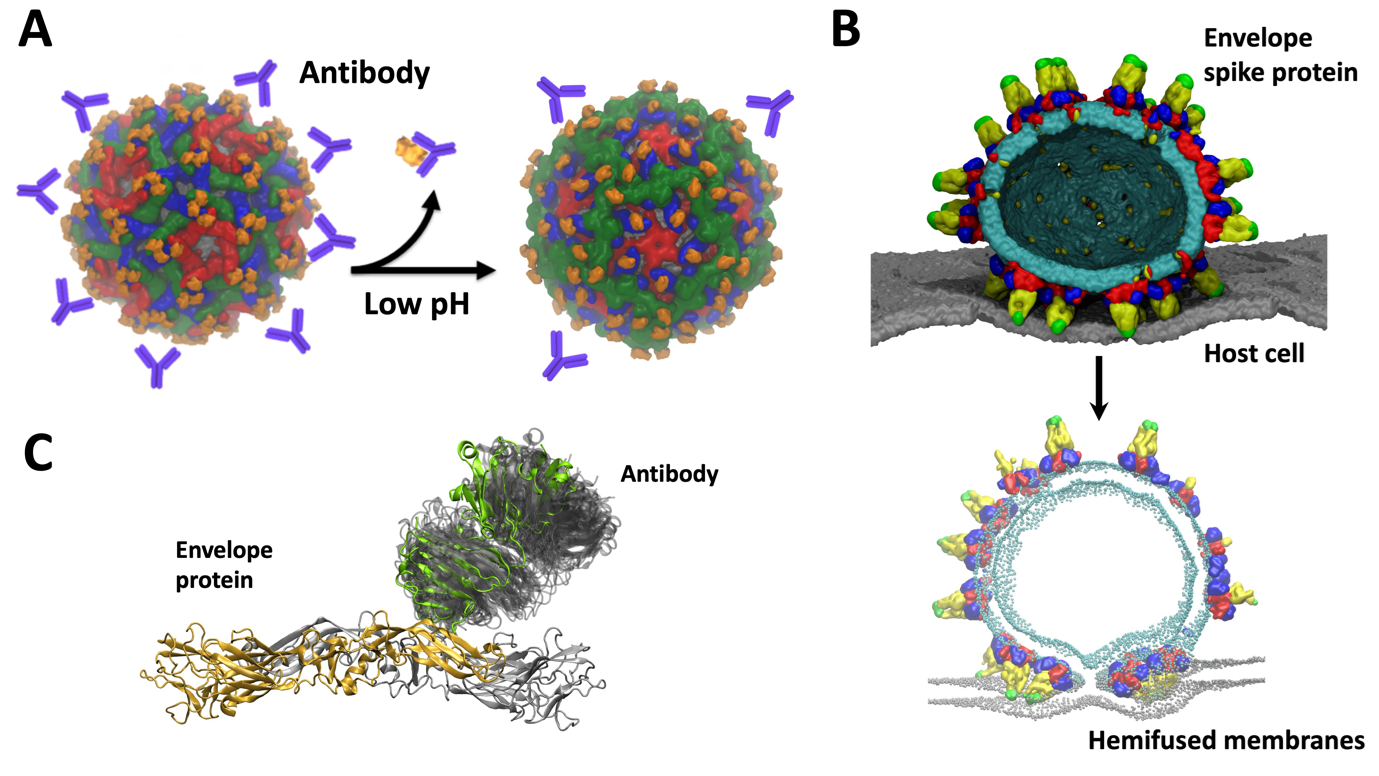
Our VLP work leverages on the previously developed in silico models of DENV in the MSMD group. These helped us to understand various aspects of the virus life cycle as a collaborative multidisciplinary project amongst several groups in Singapore. For example, consecutive infections by different serotypes of DENV can lead to “antibody dependent enhancement” (ADE) in which antibodies from a previous infection bind to the infecting virus particle but do not neutralize it. This can result in a severe case of dengue. Together with cryo-electron microscopy data from Duke-NUS, we previously explained the molecular details of ADE and suggested intermediate stages of virus maturation which opens new targets in drug design (Figure 2A). Through probing virus dynamics at the molecular level we explained the function of different viral components such as envelope-membrane interplay, envelope – host cell interactions (Figure 2B) or antibody binding (Figure 2C). We proposed new mechanisms of virus infection, replication, as well as explain the role of pH, temperature, and salt concentration which affect the virus E protein structure, a key therapeutic target.
Drug discovery is a tedious and complicate process, often with high attrition rate. To efficiently translate the fundamental knowledge to therapeutics, we combine computational modelling and simulations, in vitro and in vivo experiments to rationalize the drug development. Our current focuses are (i) development of next generation membrane active antibiotics against multi-drug resistant (MDR) bacteria; and (ii) understand the mechanism of membrane translocation of drug like molecules such as natural products, peptides etc.
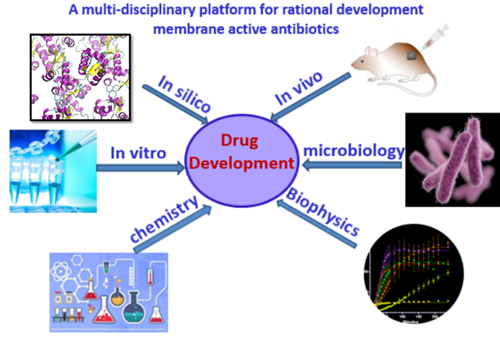
Antibiotic resistance has become a global healthcare problem due to the overuse of antibiotics. Membrane-active antimicrobials are promising to tackle the problem of rapid increases in resistance. Unlike traditional antibiotics that target specific receptors which can be easily mutated, membrane-targeting antibiotics disrupt/perturb bacterial membranes, an essential component of the bacterial cell with low mutation rate, therefore have low tendency to develop resistance. Currently we are working on two main classes of membrane-active antimicrobials, antimicrobial peptides (AMP) and small synthetic antimicrobials. We have developed a multi-disciplinary approach for rational development of membrane active antibiotics, which includes: in silico modelling and simulations, in vitro validation and characterizations and in vivo evaluations (Figure 1). This platform enables us to gain insights from different angles and has been successful for rational development of membrane active antimicrobials.
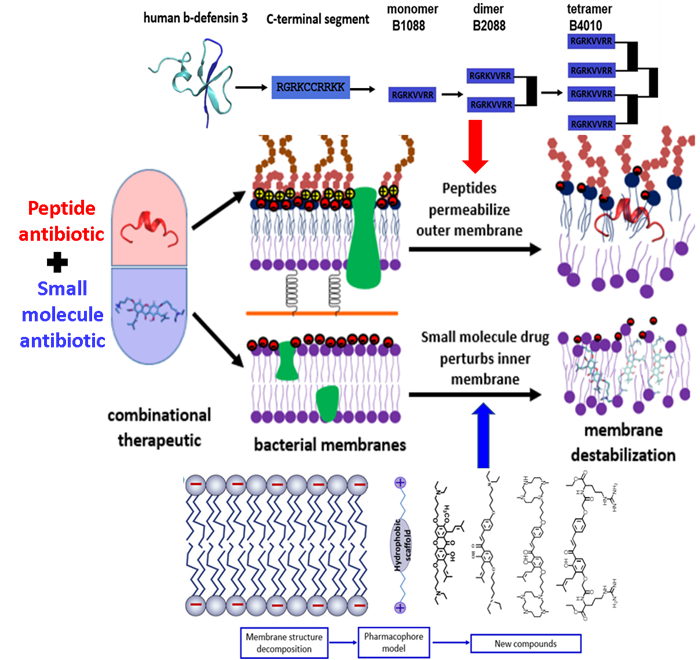
Starting from human beta-defensin 3, we have developed a series branched lipo-peptide oligomers by covalent crosslinking peptide monomer through a branched lysine core. One of the peptides B2088 display excellent antimicrobial activity against MDR bacteria yet with low toxicity. Higher oligomer such as the peptide tetramer B4010 are also active against a panel of fungi and yeasts. These peptide oligomers were found to be able to disrupt the outer membrane of Gram negative bacteria and facilitate the translocation of traditional antibiotics into the bacterial cell. On the other hand, using the strategy of fragment based drug design, for the first time, we have proposed a pharmacophore model for the design of small molecule antibiotics targeting the bacterial inner membrane. Based on the pharmacophore model, we have synthesized a small library of semi-synthetic natural products that are active against a wide range of Gram positive bacteria. By combination of the outer membrane targeting peptide antibiotics and the inner membrane targeting semi-synthetic natural products, we developed combinational therapies (Figure 2) that are active against both Gram positive and Gram negative superbugs without inducing resistance, including the recently appeared mobile colistin resistant bacteria mcr-1.
Drug delivery across the cytoplasmic membrane
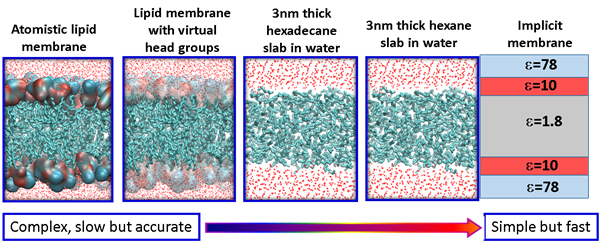
Membrane permeability is a critical factor that influences the bioavailability of a drug. Small molecule drugs generally cross the membrane via a mechanism of passive diffusion, while peptide drugs translocate across the membrane through a complex mechanism. One of our focuses is to understand the mechanism of membrane translocation of drug like molecules, including peptides and peptidomimetics using atomistic modelling and simulations. We have developed several in silico models with different level of resolutions and complexities that can be used for rapid screening of different drug molecules. For small molecule drugs, we use the atomistic lipid membrane models to accurately compute the free energy barrier along the translocation pathway. For peptides and peptidomimetics, simple membrane models are used to identify important metastable states along the translocation pathway. The final objective is to develop useful molecular parameters/descriptors that can predict the membrane permeability of drug like molecules.
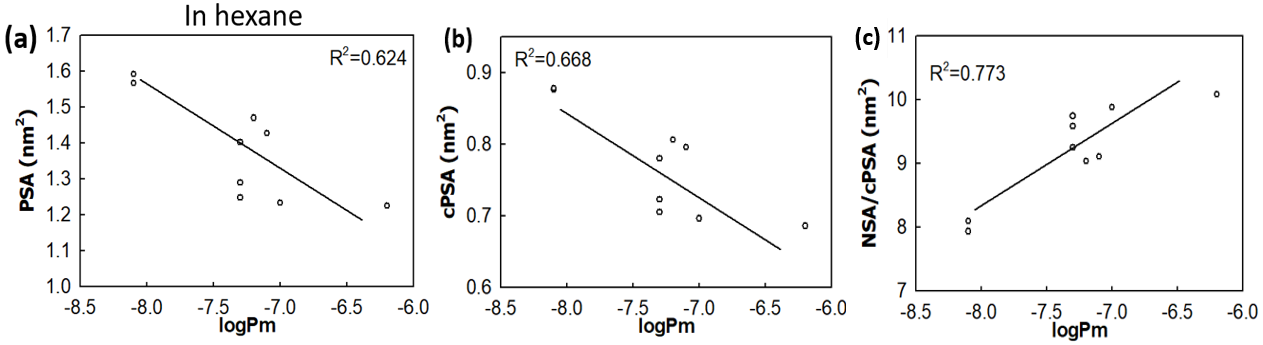
Polar surface area (PSA) is often used as a useful parameter to correlate with the membrane permeability of drug molecules. Based on the simulations a set of cyclic peptides, we have designed two new parameters: (i) the charge reweighted polar surface area cPSA=∑qi*PSAi and the ratio of non-polar surface area (NSA) to polar surface area NSA/cPSA, with the former taking the atom polarity into account and the latter considers the contribution of non-polar atoms. The two new parameters display enhanced correlation with the membrane permeability for cyclic peptides than the commonly used parameter PSA.
Our group expertise is in functional biology, chemical genetics, and high throughput screening for several phenotypic and experimental conditions. We screen the chemical libraries (small molecules, natural products, and FDA-approved drugs) to identify the interventions for aging. We combine our powerful approaches with multilayer functional analysis and identify the genes, proteins, and biological processes involved in the biology of aging and age-associated pathologies. Our group is also utilizing discovery-driven functional and chemical screening approaches to systematically identify the novel antifungal compounds and map the biological process landscapes of human fungal pathogenesis.
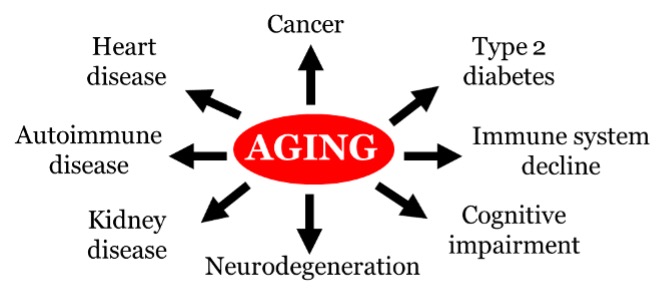
Aging population rapidly growing worldwide. Aging is the biggest risk factor for several human chronic ailments, including cancer, neurodegeneration, heart disease, diabetes, cognitive impairment and immune system decline (Figure 1). Aging is characterized by a progressive loss of physiological integrity, cellular functions and metabolic signaling. Such biological changes eventually lead to the prevalence of age-related pathologies, thereby compromise human health throughout life. Recent research in eukaryotic different model systems from single-celled yeast to nematode worms, fruit flies, mice and humans, has shown that aging mechanisms are conserved and delay aging is feasible by interventions including caloric restriction and drug administration. Our lab using budding yeast Saccharomyces cerevisiae as a model organism for study aging. The benefits of studying aging in yeast include a short generation time, a tractable life span, a genome-wide knockout library, its amenability to high-throughput functional assays and conservation of genes and pathways that uncover novel insights into human aging and interventions.
Allostery is a pervasive phenomenon whereby protein function is modulated by distant perturbations, such as binding of small molecules, mutations, and other perturbations. We investigate allosteric mechanisms using computational modeling, in conjunction with experimental data. Elucidating allosteric signaling in diverse proteins and biologics allows us to establish broadly-applicable methodologies and frameworks to modulate biomolecular functions for various purposes, including developing new therapeutic strategies and interventions.
Inducing targeted allosteric signals via ligand binding, mutation, and order-disorder transition
While the advantages of allosteric drugs in precision medicine are widely recognized, the identification of new allosteric sites, most of them have been discovered serendipitously, remains a difficult problem. Therefore, a major thread of our research is focused on in silico identification of allosteric sites and mutations in order to achieve allosteric control over protein functions.
To describe the causality and energetics of allosteric signaling, we employed the Structure-Based Statistical Mechanical Model of Allostery (SBSMMA) developed by the Berezovsky group (PLoS Comput. Biol. 12, e1004678). We introduced a method dubbed as “Reverse Perturbation”, which allows one to uncover allosteric sites by mimicking binding at functional sites (PLoS Comput. Biol. 14, e1006228). We showed that the method is able to identify known allosteric sites with high predictive power, as well as to predict latent and to design potential allosteric sites. The concept that required/desired allosteric response can be induced and fine-tuned was further proposed and demonstrated. Reverse perturbation identified distant regions containing overlapping potential allosteric sites, each with a different composition of residues and stereo-chemical properties (Figure 1, left). Subsequently, desired mode and strength of allosteric signals to targeted functional sites can be rationally induced and fine-tuned by optimizing the composition/structure of putative allosteric sites (Figure 1, right). We showed that design of new allosteric site and ligands/drugs can work via mutual adjustments to induce targeted allosteric signals - one may design/screen ligands that can bind strongly to identified allosteric sites and/or exploring suitable candidate sites that are capable of originating the needed allosteric signals for specific ligands/drugs. 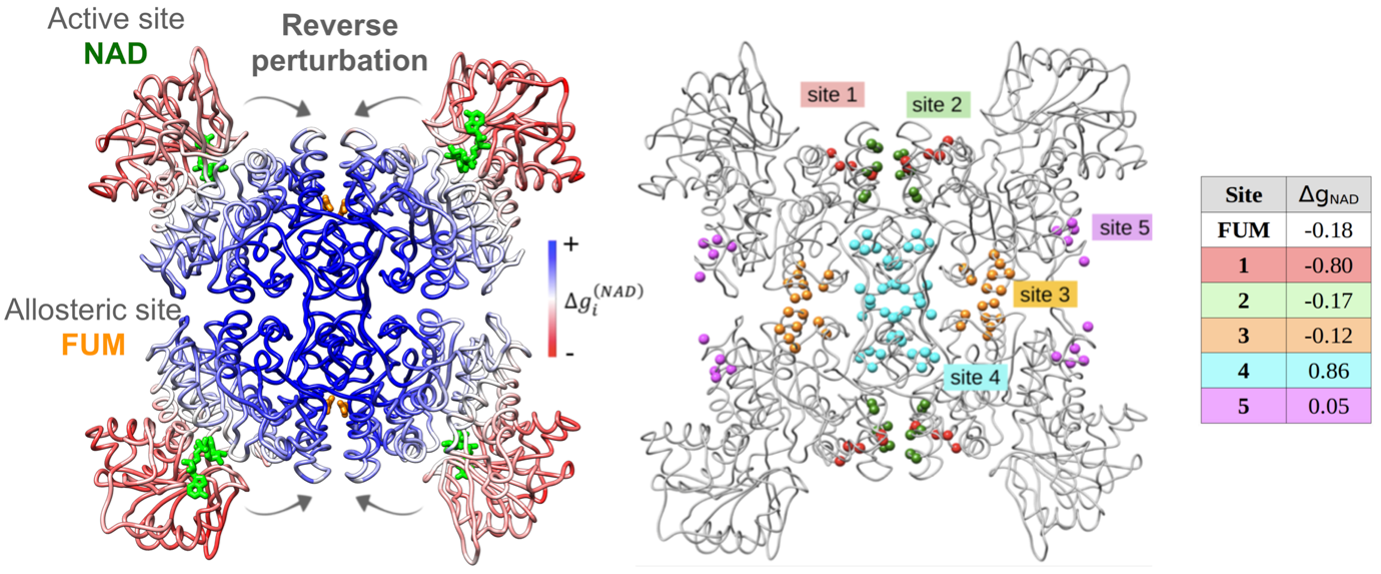
Figure 1. Left, reverse perturbation at active site NAD causes large positive free energy changes (kcal/mol) at multiple locations, including the known allosteric site FUM. Right, different arbitrarily allosteric sites within the coupled region cause varied allosteric signals in the NAD site.
It is often observed that some mutations are able to disrupt protein functions and give rise to diseases despite located far away from functional sites, suggesting they might work allosterically. We investigated the possible allosteric effects of pathological nsSNPs in a set of proteins and found that some nsSNPs can indeed lead to strong allosteric effects in the functional sites (J Mol Biol. 431, 3933). We also observed a number of uncharacterized amino acid positions able to induce similarly strong allosteric response upon mutations, which led us to introduce the concept of “Allosteric Polymorphism”. Importantly, we also identified distant amino acid substitutions counter-acting the harmful allosteric effects, illuminating their potential as rescue mutations. In another work, we found that engineering disorder-order transitions at critical positions that are allosterically coupled to functional sites allows remote modulation of protein functions (Curr Res Struc Biol. 12, 191). Altogether, these highlighted works show that ligand binding, mutations and order-disorder transition can be utilized to induce and fine-tune allosteric signaling of biologics for therapeutic and biotechnological applications. On the other hand, we have been involved in the development of online server (AlloSigMA, Nucleic Acids Res. 48, W116) and database (AlloMAPS, Nucleic Acids Res. 47, D265) by Berezovsky group, which are widely used by experimentalists and theorists world-wide for the analysis of allosteric signaling.
Conservatism and diversity in allosteric fingerprints of proteins for evolutionary-inspired engineering and design
Recognizing that a big picture view of allosteric communication would facilitate the engineering and design of allosteric control in diverse proteins and biologics, we explored the characteristics of allosteric communication in major protein folds and repeat proteins (J Mol Biol. 434: 167577). We obtained a picture of conserved allosteric communication characteristic in different fold types, modifications of the structure-driven signalling patterns via sequence-determined divergence to specific functions, as well as emergence and potential diversification of allosteric regulation in multi-domain proteins and oligomeric assemblies. Learning directly from Nature, our results will be instrumental in facilitating the engineering and de novo design of proteins with allosterically regulated functions, including development of therapeutic biologics.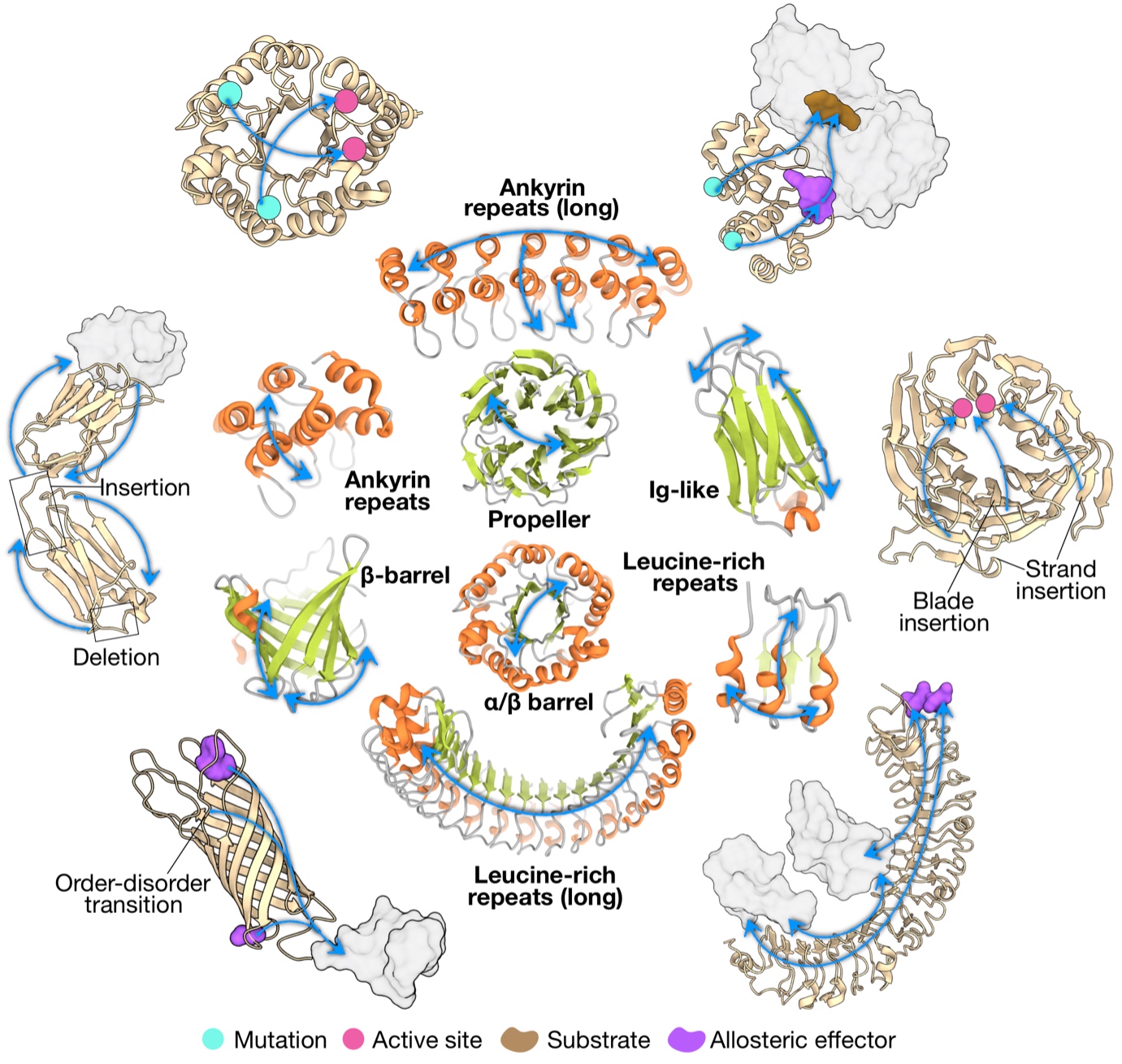
Figure 2. Archetypal patterns of allosteric communication and their potential alterations in different folds.
Towards a generic framework for designing allostery-based therapeuticsTowards a generic framework for designing
Previously developed methods on inducing targeted allosteric signaling have recently led to a broadly applicable in silico framework (Figure 3) for the analysis of allosteric signaling and the engineering and design of allosteric modulation in proteins with diverse structures/forms and functions (J. Phys. Chem. B 125, 3763).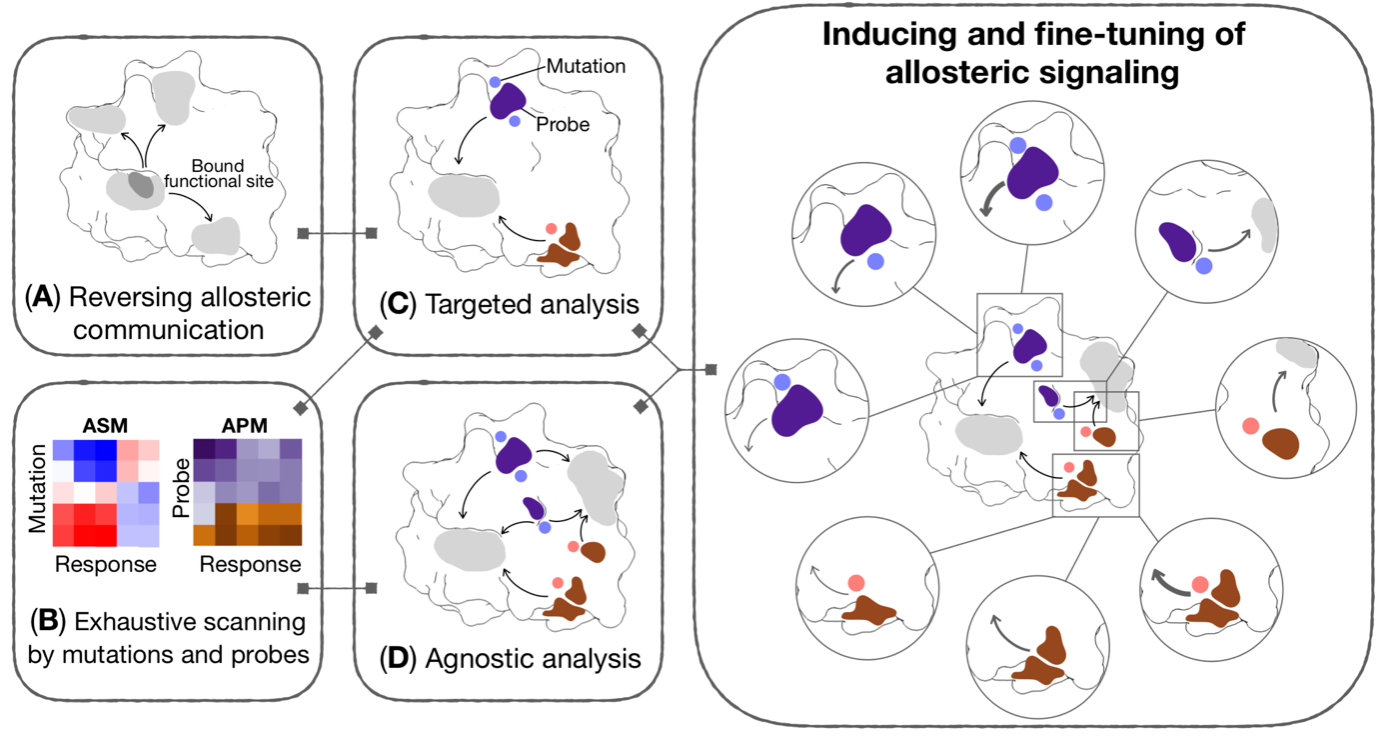
Figure 3. Generic workflow for allosteric control of protein functions and fragment-based design of allosteric sites and effectors. Different components of the workflow may be used reverse perturbation to identify coupled regions; exhaustive scanning by mutations and probes yielding comprehensive per-residue Allosteric Signaling and Probing Maps (ASM/APM); targeted and/or agnostic analyses to identify strongly modulating mutations and/or probes targeting a specific site or multiple regions of a protein, respectively.
We applied the framework to a range of human and viral proteins involved in diseases, for example isocitrate dehydrogenase causing leukemia, the Spike homotrimer and RNA polymerase from SARS-CoV-2. Using the Spike protein, the focal point of therapeutic interventions targeting COVID-19, as an example, we showed that some emerged mutations in variants of concern cause large allosteric modulation in the distant receptor binding motif (Structure 30, 590). Importantly, we also performed a comprehensive analysis of prospective druggable allosteric sites (Figure 4), and identified beneficial amino acid changes that may neutralize the deleterious allosteric effects of circulating mutations in variants of concern. As COVID-19 is transitioning into an endemic disease, our result will be important for advancing the development of allostery-based therapeutics leveraging the strength of allosteric drugs in circumventing recurring issues of drug resistance, and improving efficacies of biologics such as monoclonal antibodies and vaccines via allosteric effects of mutations.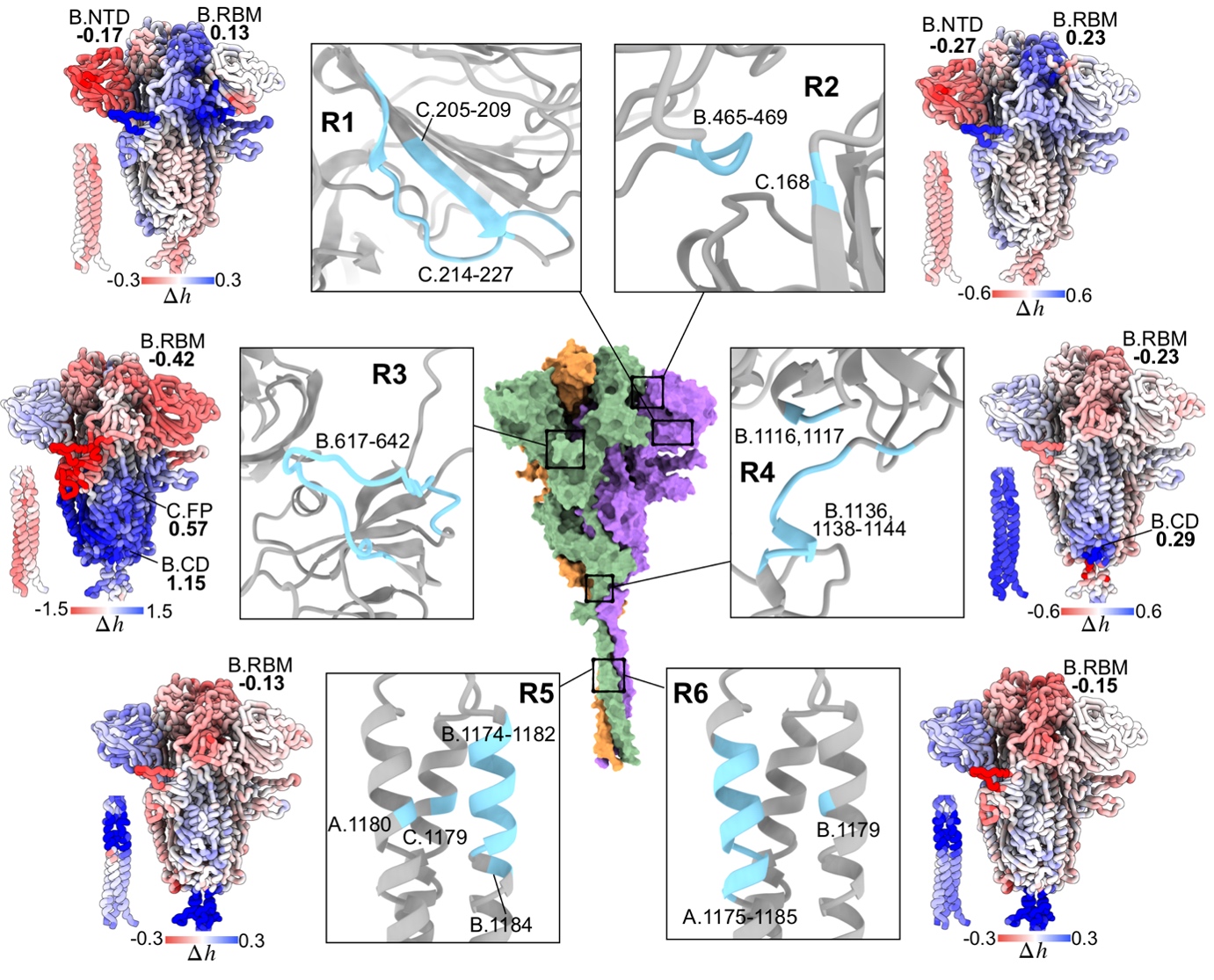
Figure 4. Identification of allosteric sites for prospective drug development targeting the spike homotrimer of SARS-CoV-2.
Old drugs in new sites - repurposing drugs to allosterically target K-Ras oncoprotein and SARS-CoV-2 proteins
Recently, we have been awarded a grant on designing allosteric drugs. Our current and upcoming projects focus on developing frameworks for tapping on multitude of existing drug libraries to identify prospective lead drugs that can be repurposed to bind allosteric sites predicted using our established methods. In a recent study, we showed that allosteric drugs and sites exhibit different physico-chemical properties compared to their orthosteric counterparts (J. Mol. Biol. 434: 167692), suggesting that one should explore the vast and considerably different chemical space in targeting allosteric sites that are typically less conserved compared to functional sites. Therefore, in parallel to drug repurposing, we are also currently utilizing fragment-based design of allosteric drugs/sites for drug development targeting K-Ras driven cancers and COVID-19.