
The broad goal of my lab is to develop computational methods to study protein-ligand interactions. The developed methods are used in basic research, to help understand and regulate biological processes, in particular, structure-function mechanisms of GPCRs, transporters, and kinases. Meanwhile, these methods were also employed in applied research, to help evaluate toxicity of chemicals including food ingredients, discover new drug leads, and engineer industrial enzymes.
COMPUTATIONAL METHOD DEVELOPMENT
We are developing computational methods to effectively and accurately model protein-ligand interactions. Two examples are given below on chemical toxicity prediction and enzyme engineering.
Chemical Toxicity Prediction
We constructed an in-silico platform for chemical toxicity prediction (Figure 1). Using ToxCast data of 12 nuclear receptors, we systematically analysed how multiple crystal protein structures can be used in screening protocols to distinguish actives from inactives. The methods used for data fusion are consensus docking scores from multiple conformations at a single functional state (agonist-bound or antagonist-bound), multiple functional states, and multiple pockets (orthosteric and allosteric sites). The results suggested that such a consensus approach could have a greater discriminating power between active and inactive molecules with respect to the standard single-structure based approach. Currently we are improving this structure-based approach with the aid of AI.
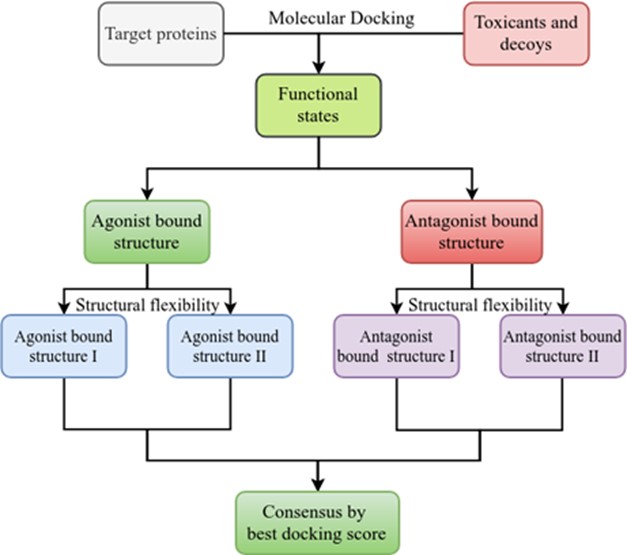
Figure 1. The in-silico platform for chemical toxicity prediction, applied to nuclear receptors.
Structure-AI method for Enzyme Engineering
We are collaborating with BII, ISCE2 and IHPC colleagues, developing “an Integrative Computational/Experimental Workflow for Enzyme Development”, to improve the speed of enzyme engineering and to build a panel of enzymes with activity against industrially relevant panel of substrates. My lab has developed a computational method that integrates machine learning and deep learning models with conventional bioinformatics for enzyme engineering. This new method has shown > 90% accuracy when it was used to predict better mutants of naturally occurring galactose oxidase (GAOx) in terms of industry-related substrate activity, and to predict new substrates for selected GAOx out of industry-related chemical databases, with improved speed of evolution and faster biocatalyst development that are critical for manufacturing processes and of high industrial importance (Figure 2). In collaboration with Dr. Wen Shan Yew’s lab at NUS, we also successfully applied the computational method to the engineering of NphB produces the cannabinoid cannabigerolic acid (CBGA), the key precursor to many cannabinoids.
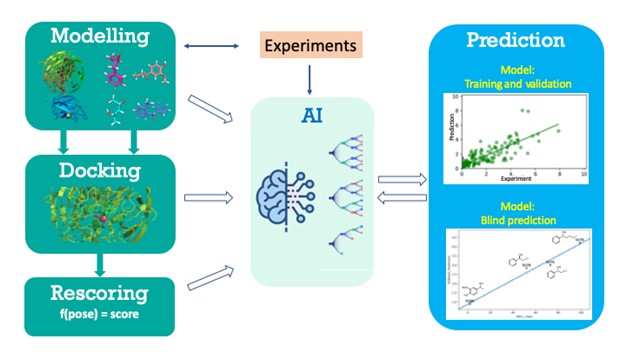
Figure 2. The computational method that integrates machine learning models with conventional bioinformatics for enzyme engineering, applied to galactose oxidase.
GPCR & Kinase: Structure-function Mechanisms
GPCRs play important roles in cell signaling pathways. Their dysfunction causes many human developmental and metabolic disorders, as well as certain cancers. In collaboration with Dr. Cheng Zhang from the University of Pittsburgh, we contributed to the determination of crystal structures of GPCRs including a lipid GPCR CRTH2 as the receptor for prostaglandin D2, the C5a receptor (C5aR) that can induce strong inflammatory events in response to the anaphylatoxin C5a peptide, and N-formyl peptide receptor 2 (FPR2) that is a high affinity receptor for the arachidonic acid metabolites. We further studied C5aR, CRTH2, and GPR84 with MD simulations, revealing a distinctive lipid recognition mechanism for CRTH2, a cooperative two-site binding mechanism for C5aR, and the molecular basis for the high selectivity of GPR84 for MCFAs as well as its potential routes of ligand binding and dissociation. Currently we are working towards the identification of novel orthosteric and allosteric modulators and better understanding of structure-function mechanisms, for other lipid GPCRs (Figure 3).
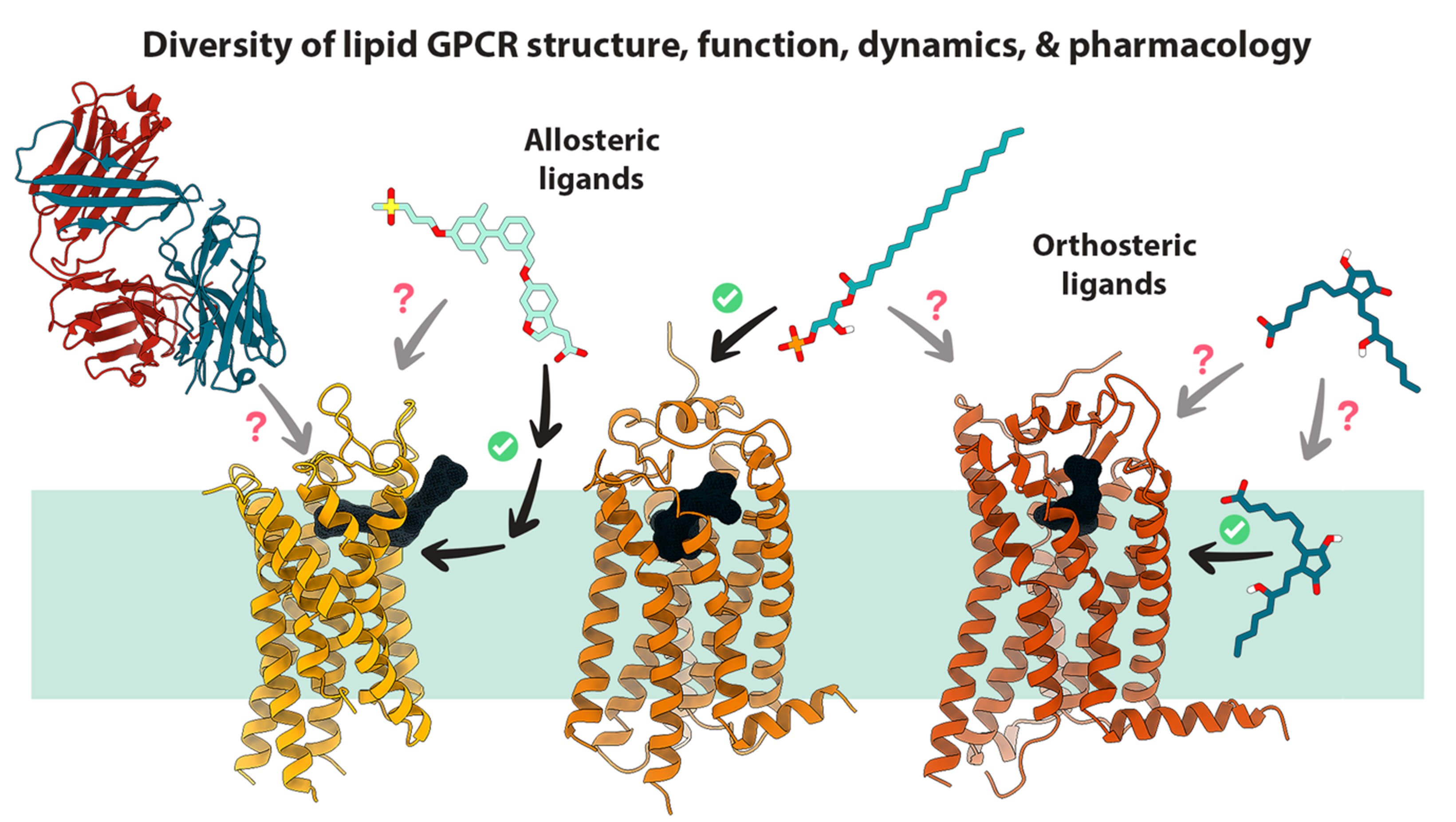
Figure 3. Recent advances in structure, function, dynamics, and pharmacology of lipid GPCRs.
Kinases play critical roles in cell signaling pathways associated with cell survival, growth, differentiation, proliferation, and migration. Together with Dr. Jiancheng Hu at NCCS, we have, for the first time, demonstrated that the stability of R-spine, a typical hydrophobic architecture in active kinase, determines the resistance of oncogenic BRAF mutants to inhibitors (Figure 4). Our preliminary results also suggested that a similar mechanism might apply to EGFR mutations. On this basis, we are developing computational methods to predict drug sensitivity of kinase mutations in cancers, and innovative precision medicines against cancers harboring specific kinase mutations.
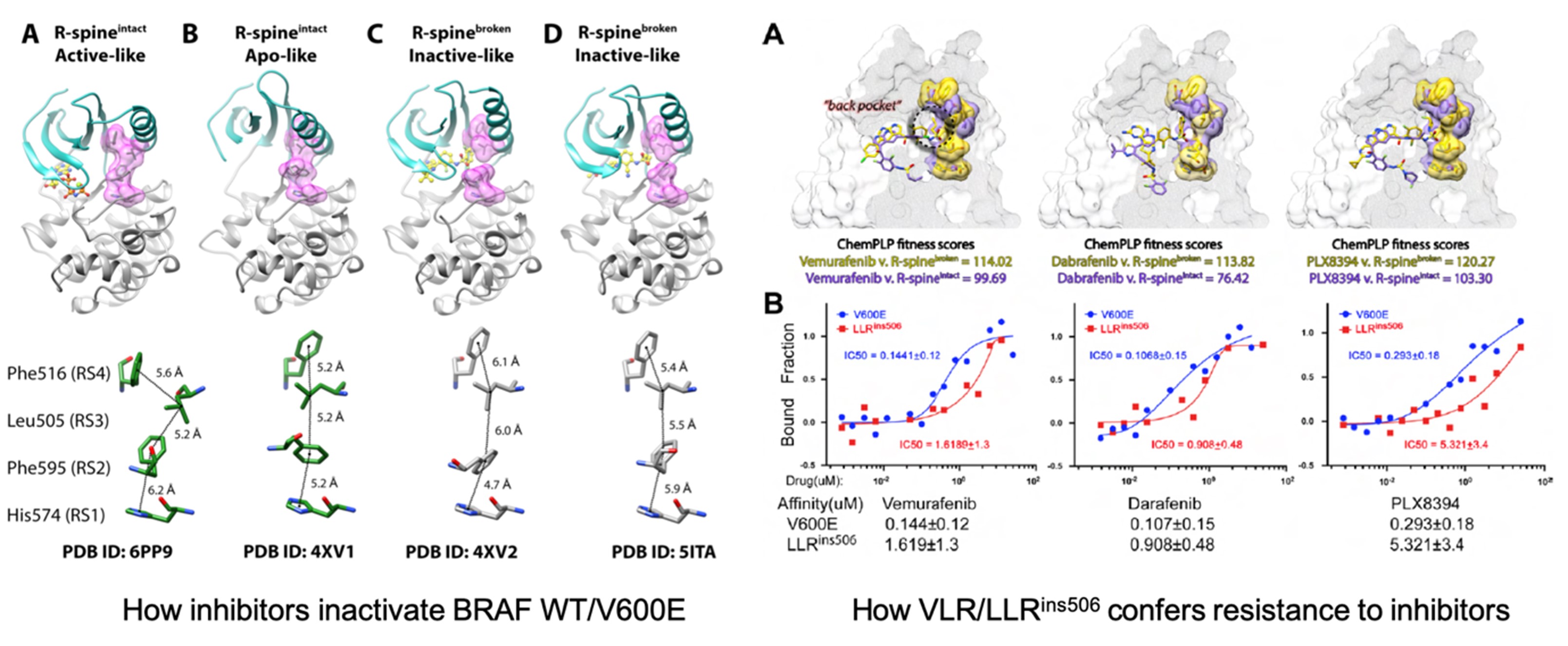
Figure 4. BRAF R-spine configurations and drug responses.
| Senior Principal Investigator | FAN Hao | [View Bio] |
| Senior Scientist | VERMA Ravi Kumar |
| Senior Scientist (Collaborator) | LIN Fu |
| Research Scientist (Adjunct) | KRISHNA DEEPAK R.N.V. |
| Senior Scientist | JALADANKI Chaitany`a Kumar |
| Senior Scientist | SUPEKAR Shreyas |
| Post-Doctoral Research Fellow (Collaborator) | HARTONO Yossa Dwi |
| Post-Doctoral Research Fellow (Collaborator) | BARBIERI Alessandro |
| Scientist | WEI Wan |
| Research Officer | RAYAKAR Achal Ajeet |
| PhD Student | BEHN Julian |
| PhD Student (co-supervisor) | WANG Kaichen |
 | FAN Hao Senior Principal Investigator Email: fanh@a-star.edu.sg Research Group: Structure-based Ligand Discovery and Design |
Dr. Hao Fan received his undergraduate degree in Biological Sciences in University of Science and Technology of China (USTC). He obtained his PhD in Biophysical Chemistry in Dr. Alan Mark’s lab at University of Groningen (RUG). He worked as postdoctoral fellow followed by research scientist in both Dr. Andrej Sali’s lab and Dr. Brian Shoichet’s lab at University of California, San Francisco (UCSF). He was appointed Principal Investigator at the Bioinformatics Institute (BII), A*STAR since 2014. Currently he is Adjunct Associate Professor at Signature Research Programme in Cancer & Stem Cell Biology (“CSCB”) and Centre for Computational Biology (“CCB”), Office of Research in Duke-NUS Medical School (“Duke-NUS”).
| Senior Scientist | VERMA Ravi Kumar |
| Senior Scientist (Collaborator) | LIN Fu |
| Research Scientist (Adjunct) | KRISHNA DEEPAK R.N.V. |
| Senior Scientist | JALADANKI Chaitany`a Kumar |
| Senior Scientist | SUPEKAR Shreyas |
| Post-Doctoral Research Fellow (Collaborator) | HARTONO Yossa Dwi |
| Post-Doctoral Research Fellow (Collaborator) | BARBIERI Alessandro |
| Scientist | WEI Wan |
| Research Officer | RAYAKAR Achal Ajeet |
| PhD Student | BEHN Julian |
| PhD Student (co-supervisor) | WANG Kaichen |