
Our group studies structural and functional motifs in RNA. We use a wide range of techniques to identify and characterize different types of RNA using sequence analysis, annotation, analysis of chemical structure probing and crosslinking data, secondary and tertiary structure prediction, molecular modelling, AI-based modelling methods, and molecular dynamics simulations. Our primary focus are viral genomic and subgenomic RNAs, as well as RNAs in other pathogenic processes including bacteria and cancers. We analyse RNA structure, RNA-RNA and RNA-Protein interactions and how these interactions affect diseases. Systems of interest span gastric cancer, mosquito-borne infections, Enteroviruses, Influenza, as well as marine pathogens. We are working to identify regulatory networks at the RNA level throughout human cell differentiation. Finally, we are studying the factors that influence manufacturability of RNAs for therapeutic use.
Most pandemics of recent decades can be traced to RNA viruses, including HIV, SARS, influenza, dengue, Zika, and SARS-CoV-2. These RNA viruses impose considerable social and economic burdens on our society. As these RNA viruses utilize an RNA genome, which is important for different stages of the viral life cycle, including replication, translation, and packaging, studying which folds the genome adopts at different stages is important to understand virus function. We employ techniques combining computational and high-throughput RNA structure-mapping approaches to aid in the understanding of structures within RNA virus genomes. Over the last year we made several advances in our understanding of flavivirus biology and the role the RNA genome of these viruses plays in their pathogenicity. We were able to show that specific RNA-RNA interactions with host transcripts can have pro- and antiviral effects and structural variants, even in UTRs, modulate the fitness of dengue virus. (1) RNA structure plays a crucial role in viral fitness and disrupting such structure creates attenuated viruses purely through synonymous mutations. This attenuation is stable in mosquitos and human hosts. (2) Finally, viral capsid proteins and their interactions with viral genomic RNA are crucial for packaging. Interestingly, binding sites of capsid proteins coincides with sites forming extensive RNA-RNA interactions in virions. We thus postulate that capsid protein is a key driver of the compaction the genomic RNA needs to undergo when getting loaded into nascent viral particles. (3)
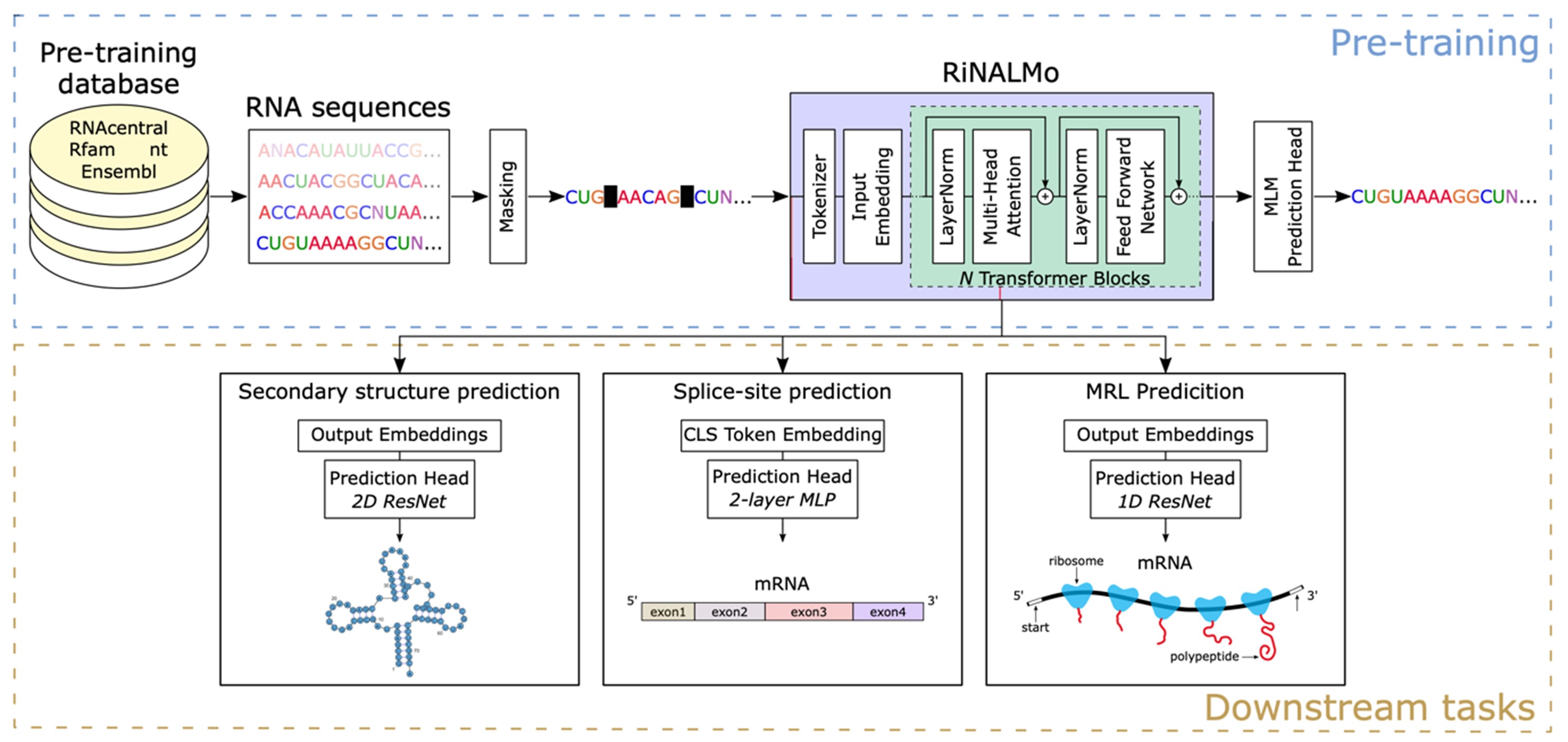
Figure 1. RiNALMo pre-training and applications. In the pre-training stage, RiNALMo is trained on unlabeled RNA sequences from several databases using masked language modeling (MLM). The final language model comprises 33 Transformer blocks with the embedding dimension of 1280. Once pre-trained, RiNALMo is separately fine-tuned for several structural and functional downstream tasks in which its expressive output embeddings, utilized by the prediction heads, significantly improve the performance.
We investigated the RNA structure and RNA-RNA interactions of wildtype (WT) and a variety of variants of concern including alpha, beta, delta and omicron strains of SARS-CoV-2 in cells using Illumina and Nanopore platforms. We identify a number of functional structural elements within the SARS-CoV-2 genome. Proximity ligation sequencing allowed us to identify hundreds of RNA-RNA interactions within the virus genome and between the virus and host RNAs. We investigated the structural effect of single mutations, their impact on RNA-binding protein associations and long-range RNA-RNA interactions. Structure in the SARS-CoV-2 genome is concentrated into highly conserved structure hubs. These structure hubs show more local secondary structure, more long-range interactions within the genome, more interactions with host transcripts and more alternative interactions for the same sequence position.
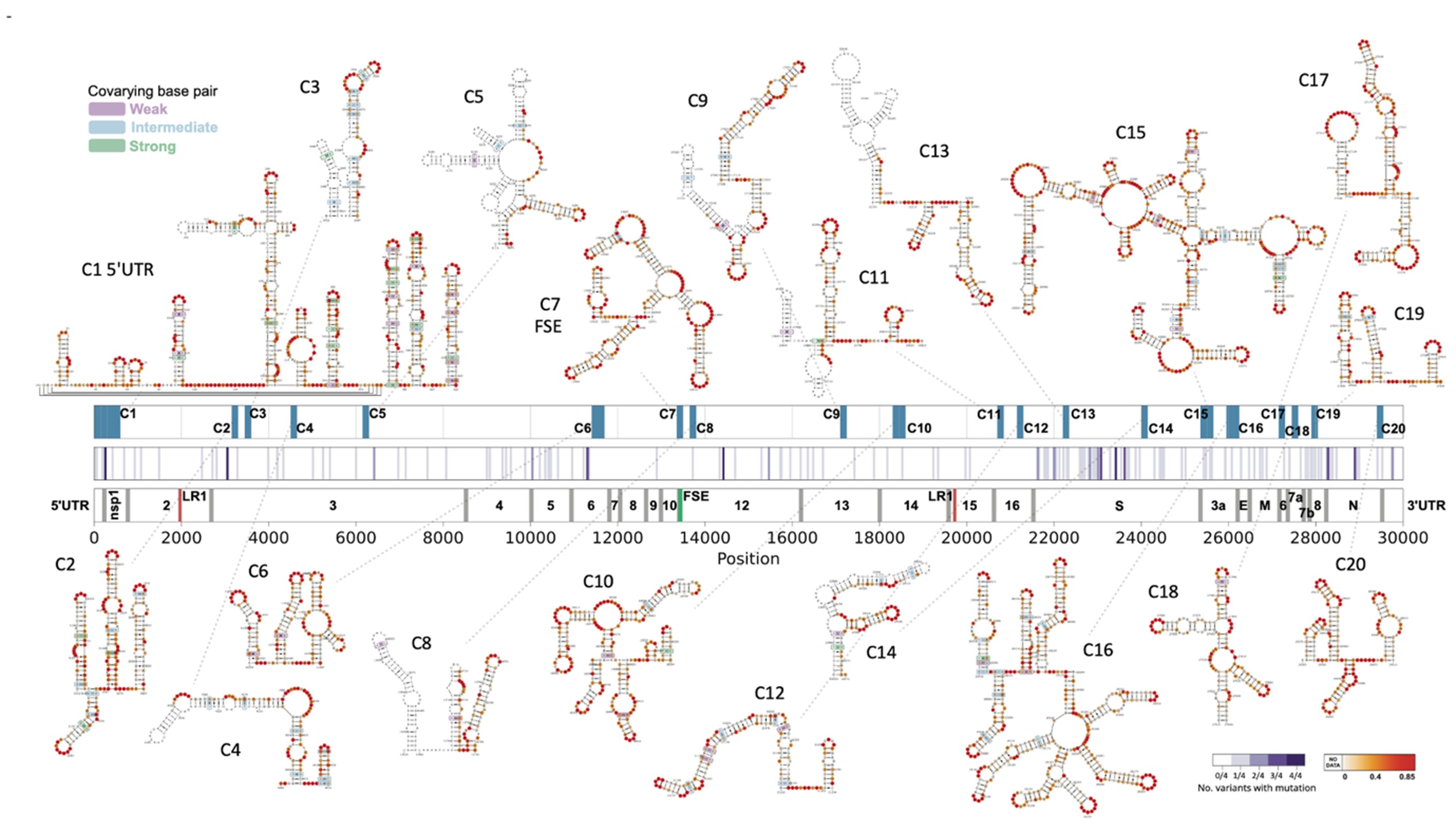
Figure 2. SARS-CoV-2 shows a number of highly conserved structural motifs. We were able to confirm the functional role of many of these motifs through mutagenesis experiments. Interestingly, structured elements cluster in hubs that are enriched not only in local structure, but also play a key role in long-range RNA-RNA interactions within the viral genome as well as in RNA-RNA interactions of the viral genome with host transcripts. These structure hubs are distributed throughout the genome and are interspersed with regions exhibiting relative structural paucity.
RNA structure is critical for multiple steps in gene regulation. However, how the structures of transcripts differ both within and between individual cells is unknown. Here we develop a SHAPE-inspired method called single-cell structure probing of RNA transcripts that enables simultaneous determination of transcript secondary structure and abundance at single-cell resolution. We applied single-cell structure probing of RNA transcripts to human embryonic stem cells and differentiating neurons. Remarkably, RNA structure is more homogeneous in human embryonic stem cells compared to neurons, with the greatest homogeneity found in coding regions. More extensive heterogeneity is found within 3′ untranslated regions and correlates with specific RNA-binding proteins. Overall, RNA structure profiles better discriminate cell type and differentiation stage than gene expression profiles alone. (6)
| Principal Investigator | HUBER Roland G. | [View Bio] |
| Senior Scientist | DEFALCO Louis |
| Senior Scientist | KULKARNI Mandar |
| Research Officer | CHIAM Aryeh Joseph |
 | HUBER Roland G. Principal Investigator Email: rghuber@bii.a-star.edu.sg Research Group: Function and Structure of RNA |
Dr. Roland G. Huber studied computational biology at the University of Innsbruck and at Yale University. After obtaining his PhD he commenced postdoctoral research at A*STAR in Singapore and was appointed as principal investigator in 2019. His group at the Bioinformatics Institute is primarily interested in the interplay of structure and function of RNA in gene regulation and infectious disease using statistical modeling, multi-omics data, and molecular simulations to reveal the structures and functional mechanisms of folded RNA and RNA-protein complexes. The group has previously worked on a variety of tropical and emerging infectious diseases including Dengue, Zika, and SARS-CoV-2. Other areas of interest are multi-omics and metagenomic analysis, particularly in the fields of animal health & aquaculture, and human immunology & ageing
| Senior Scientist | DEFALCO Louis |
| Senior Scientist | KULKARNI Mandar |
| Research Officer | CHIAM Aryeh Joseph |