
Our group expertise is in computational protein sequence and structure analysis to predict various aspects of molecular and cellular functions (enzymatic activities, posttranslational modifications, cleavage, translocation signals, 3D structures, effects of mutations, phylogenetic relationships, cellular pathways etc.) for discovering the molecular mechanisms of biological and clinical phenotypes and experimental validation together with collaborators. Our repertoire of computational analysis methods is applicable and useful in multiple research areas but our main focus currently is on infectious diseases, human mutations, allergy and enzyme function prediction.
One of our traditional strongholds since the swine flu in 2009 is infectious disease research. Our FluSurver (https://flusurver.bii.a-star.edu.sg/) is the most complete one-stop influenza mutation analysis tool being used by researchers and surveillance experts globally. We have several published and ongoing projects with the WHO CC in Australia and National Influenza Centres relating to influenza drug resistance, viral fitness, host specificity and antigenic changes. The FluSurver is also a primary analysis tool for the global data science initiative GISAID, the most complete influenza database used by WHO flu surveillance networks. Our most recent FluCluster-AI tool allows near real-time surveillance and visualization of Influenza variants that predicts antigenic drifts, growth rates as well as automatic detection of variant correlations with clinical and epidemiological data (Figure 1). BII/A*STAR has been a technical and scientific partner to GISAID for several years providing analysis tools.
When the new coronavirus was discovered by Chinese colleagues, the sequences were shared using GISAID’s platform on January 10th 2020 (https://gisaid.org). Since it was a new virus, this required a new database system to be designed and built, as well as a new workflow to curate and release sequences. GISAID called on the Singapore team to help. In addition to the expert advice contributing to build the new system and annotation tools like the CoVsurver (https://gisaid.org/covsurver), the Singapore team has been critical in processing the incoming genomes with quality checks and analysis reports since the beginning and was reinforced soon by more international colleagues. This has enabled global genome sharing from the first day to over 17 million genomes from 215 countries and territories by 2024, earning the former WHO Chief Scientist’s commendation of GISAID as a “game changer”. (Swaminathan, Nature 2020). This had significant impact for Singapore and the world. Diagnostics, drugs, and vaccine development were started based on sequences in GISAID and are constantly checked with new incoming data if they are still working well and help to identify new variants. In Singapore, BII has become a hub for multiple institutions and agencies to access and benefit from the GISAID work, from other A*STAR colleagues to NPHL/NCID, MOH, Duke-NUS, hospitals and DSO.
Because we can quickly go from genomes to protein structures through modelling in our computers often only requiring the new sequences as input, our group offers powerful support in infectious disease surveillance and rapid outbreak investigations to get a quick handle on bugs here and around the world. Besides Influenza and hCoV-19, we also helped characterizing Mpox, MERS, Ebola, HIV, Noro, Adeno, RSV, Hepatitis C, West Nile, Dengue, Zika, Chikungunya and Oropouche viruses. Through close collaboration with the National Public Health Laboratory at the National Centre for Infectious Diseases of the Ministry of Health we contribute our knowledge and computational expertise at the national frontline for infectious disease surveillance.
We aim at bridging the gap from nucleotide variation to protein structures to interpret effects of human mutations. For example, we have helped clinical collaborators to analyze variants found in patients and tried to mechanistically explain their possible role in a range of diseases like cancer, myopia, leprosy or atopic dermatitis. We are participating in the National Precision Medicine Programme to help mapping mutations into 3D protein structures relative to drug binding sites supporting our colleagues at GIS, LKC and PRECISE. Multinational Procter & Gamble (P&G) and BII have jointly developed animal-testing-free Bioinformatics techniques for assessing the allergy potential of proteins using their amino acid sequence and tertiary structure (https://research.a-star.edu.sg/articles/highlights/predicting-protein-allergens-accurately/ ) with AllerCatPro (https://allercatpro.bii.a-star.edu.sg/). Further, in collaboration with P&G, we have recently gained valuable insights into enhancing allergenicity potentialanalysis for enzyme families. The results of these studies will help lay the foundation for extending the in silico tool to assess the allergenicity of industrially used enzymes. Our team, together with the Singapore Institute of Food and Biotechnology Innovation (SIFBI), NTU FRESH and James Cook University (JCU), apply AllerCatPro to the safety assessment of proteins found in novel foods, such as those replacing meat with alternative protein sources including as advisor to the Singapore Food Authority. Through a Singapore Food Agency funded project, we collaborate with SIgN, NTU and JCU to evaluate food safety risks associated with innovative solutions aimed at meeting and securing Singapore’s nutritional needs through domestic fish production from sustainable urban aquaculture. Since 2023, we have also created a technology platform (https://www.rsc.a-star.edu.sg/technologyplatforms/scientific-side-menu/scientific-information/food-and-consumer-care/assessment-of-protein-allergenicity-potential) dedicated to protein safety assessment using AllerCatPro, offering a consultancy package for industries to predict potential allergens in novel foods. In other notable projects with SIFBI and other institutes often including industry collaborations, we are applying our sequence function and pathway analysis capabilities to support the Natural Product Library, Biotransformation and Synthetic Biology programmes as well as the Pharma Innovation Programme Singapore.
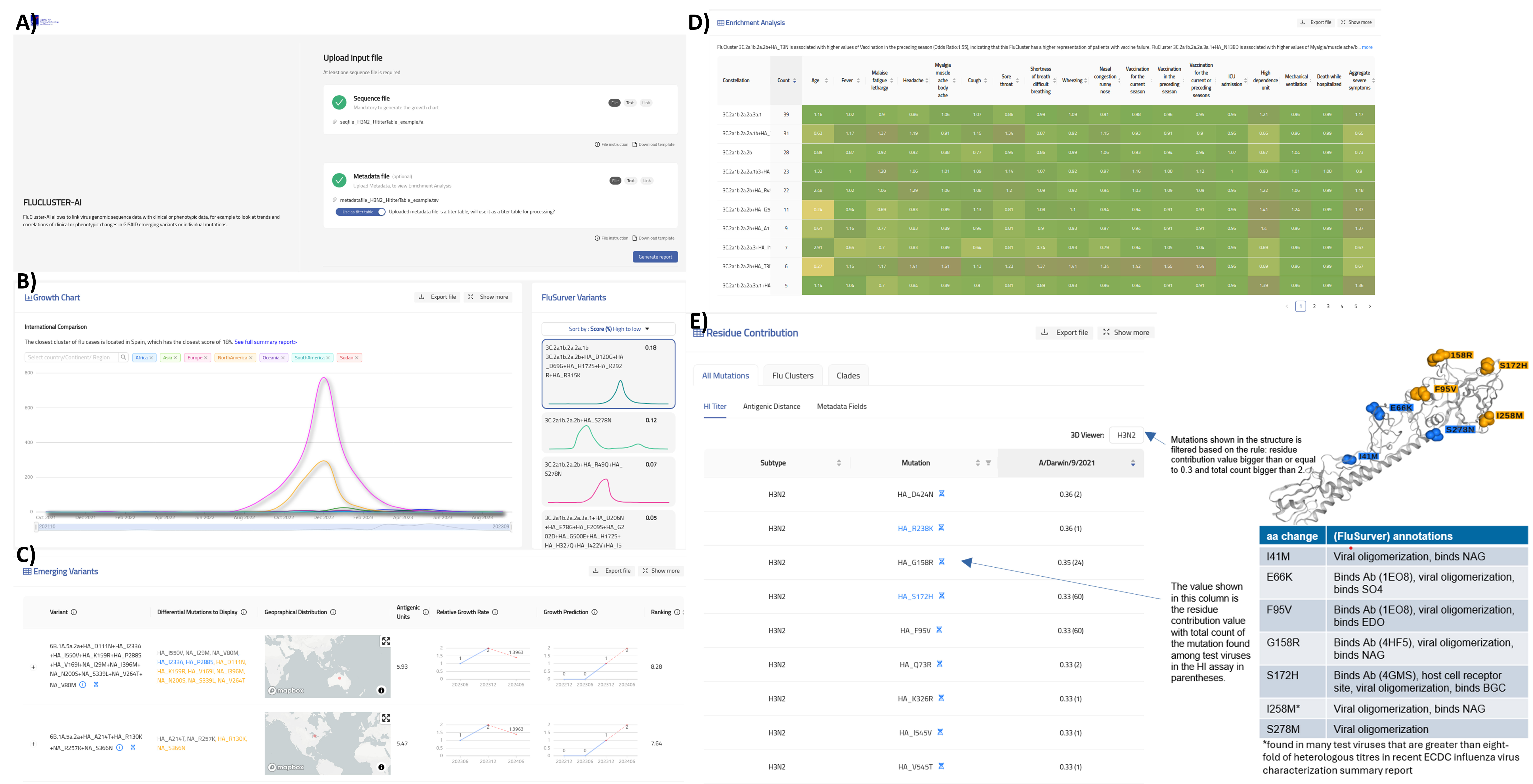
Figure 1. Our FluCluster-AI tool at GISAID. A) Landing page where users upload their influenza sequences and their associated clinical or epidemiological metadata. B) Growth trends of influenza variants that are closest to users input sequences. C) Most recent emerging FluClusters and their predicted growth and antigenic distances. D) Odds Ratio association analysis of FluClusters to each of the metadata provided by the user. E) Individual amino acid residue contribution to each of the metadata provided by the user. FluCluster-AI can also process raw HI-titer tables and highlight to users residue changes that contribute most significantly to antigenic distances and display them in 3D structure.
| Executive Director | MAURER-STROH Sebastian | [View Bio] |
| Principal Scientist | LIMVIPHUVADH Vachiranee |
| Senior Scientist | KENANOV Dimitar |
| Senior Scientist | MAK Tze Minn Sandy |
| Senior Scientist | CHIA Jing Ting Charmaine |
| Senior Scientist | WAILAN Alexander |
| Scientist | CHONG Cheng Shoong Ken |
| Principal Research Officer | LEE Tze Chuan Raphael |
| Lead Research Officer | XU Yani Angela |
| Senior Research Officer | CHEW Yi Hong |
| Senior Research Officer | NG Ting Ting |
| Research Officer | MAKHEJA Meera |
| PhD Student | ATTIQUE Syed Awais |
Li M, Ang KS, Teo B, Rom U, Nguyen MN, Maurer-Stroh S, Chen J. Rediscovering publicly available single-cell data with the DISCO platform. Nucleic Acids Res. 2024 Nov 13:gkae1108. doi: 10.1093/nar/gkae1108. Epub ahead of print. PMID: 39535037.
Getchell M, Wulandari S, de Alwis R, Agoramurthy S, Khoo YK, Mak TM, Moe L, Stona AC, Pang J, Momin MHFHA, Amir A, Andalucia LR, Azzam G, Chin S, Chookajorn T, Arunkumar G, Hung DT, Ikram A, Jha R, Karlsson EA, Le Thi MQ, Mahasirimongkol S, Malavige GN, Manning JE, Munira SL, Trung NV, Nisar I, Qadri F, Qamar FN, Robinson MT, Saloma CP, Setk S, Shirin T, Tan LV, Dizon TJR, Thayan R, Thu HM, Tissera H, Xangsayarath P, Zaini Z, Lim JCW, Maurer-Stroh S, Smith GJD, Wang LF, Pronyk P; Asia Pathogen Genomics Initiative (Asia PGI) consortium. Pathogen genomic surveillance status among lower resource settings in Asia. Nature Microbiol. 2024 Oct;9(10):2738-2747. doi: 10.1038/s41564-024-01809-4. Epub 2024 Sep 24.
Sakai A, Singh G, Khoshbakht M, Bittner S, Löhr CV, Diaz-Tapia R, Warang P, White K, Luo LL, Tolbert B, Blanco M, Chow A, Guttman M, Li C, Bao Y, Ho J, Maurer-Stroh S, Chatterjee A, Chanda S, García-Sastre A, Schotsaert M, Teijaro JR, Moulton HM, Stein DA. Inhibition of SARS-CoV-2 growth in the lungs of mice by a peptide-conjugated morpholino oligomer targeting viral RNA. Mol Ther Nucleic Acids. 2024 Sep 10;35(4):102331. doi: 10.1016/j.omtn.2024.102331. PMID: 39376996; PMCID: PMC11456799.
Goh YS, Fong SW, Hor PX, Loh CY, Wang B, Salleh SNM, Ngoh EZX, Lee RTC, PohXY, Rao S, Chia PY, Ong SWX, Lee TH, Lim C, Teo J, Pada S, Sun LJ, Ong DLS, Somani J, Lee ES, Maurer-Stroh S, Wang CI, Leo YS, Lye DC, Young BE, Ng LFP, Renia L; NCID Study Group; COVID-19 Cohort Study Group. Variant-Specific IgA Protects Against Omicron Infection. J Infect Dis. 2024 Aug 16;230(2):e287-e291. doi: 10.1093/infdis/jiad525. PMID: 37996071; PMCID: PMC11326848.
Karnaneedi S, Johnston EB, Bose U, Juhász A, Broadbent JA, Ruethers T, Jerry EM, Kamath SD, Limviphuvadh V, Stockwell S, Byrne K, Clarke D, Colgrave ML, Maurer-Stroh S, Lopata AL. The Allergen Profile of Two Edible Insect Species-Acheta domesticus and Hermetia illucens. Mol Nutr Food Res. 2024 Aug;68(16):e2300811. doi: 10.1002/mnfr.202300811. Epub 2024 Jul 18. PMID: 39022859.
Selvarajoo K, Maurer-Stroh S. Towards multi-omics synthetic data integration. Brief Bioinform. 2024 Mar 27;25(3):bbae213. doi: 10.1093/bib/bbae213. PMID: 38711370; PMCID: PMC11074586.
Krutz NL, Kimber I, Winget J, Nguyen MN, Limviphuvadh V, Maurer-Stroh S, Mahony C, Gerberick GF. Identification and semi-quantification of protein allergens in complex mixtures using proteomic and AllerCatPro 2.0 bioinformatic analyses: a proof-of-concept investigation. J Immunotoxicol. 2024 Dec;21(1):2305452. doi: 10.1080/1547691X.2024.2305452. Epub 2024 Jan 31. PMID: 38291955.
Karnaneedi S, Limviphuvadh V, Maurer-Stroh S, Lopata AL. De Novo Transcriptomic Analyses to Identify and Compare Allergens in Foods. Methods Mol Biol. 2024;2717:351-365. doi: 10.1007/978-1-0716-3453-0_24. PMID: 37737997.
Gangavarapu K, Latif AA, Mullen JL, Alkuzweny M, Hufbauer E, Tsueng G, Haag E, Zeller M, Aceves CM, Zaiets K, Cano M, Zhou X, Qian Z, Sattler R, Matteson NL, Levy JI, Lee RTC, Freitas L, Maurer-Stroh S; GISAID Core and Curation Team; Suchard MA, Wu C, Su AI, Andersen KG, Hughes LD. Outbreak.info genomic reports: scalable and dynamic surveillance of SARS-CoV-2 variants and mutations. Nature Methods. 2023 Apr;20(4):512-522. doi: 10.1038/s41592-023-01769-3. Epub 2023 Feb 23. PMID: 36823332.
Wong E, Bertin N, Hebrard M, Tirado-Magallanes R, Bellis C, Lim WK, Chua CY, Tong PML, Chua R, Mak K, Lim TM, Cheong WY, Thien KE, Goh KT, Chai JF, Lee J, Sung JJ, Wong TY, Chin CWL, Gluckman PD, Goh LL, Ban KHK, Tan TW; SG10K_Health Consortium; Sim X, Cheng CY, Davila S, Karnani N, Leong KP, Liu J, Prabhakar S, Maurer-Stroh S, Verma CS, Krishnaswamy P, Goh RSM, Chia I, Ho C, Low D, Virabhak S, Yong J, Zheng W, Seow SW, Seck YK, Koh M, Chambers JC, Tai ES, Tan P. The Singapore National Precision Medicine Strategy. Nature Genet. 2023 Feb;55(2):178-186. doi: 10.1038/s41588-022-01274-x. Epub 2023 Jan 19. PMID: 36658435.
Brito AF, Semenova E, Dudas G, Hassler GW, Kalinich CC, Kraemer MUG, Ho J, Tegally H, Githinji G, Agoti CN, Matkin LE, Whittaker C; Bulgarian SARS-CoV-2 sequencing group; Communicable Diseases Genomics Network (Australia and New Zealand); COVID-19 Impact Project; Danish Covid-19 Genome Consortium; Fiocruz COVID-19 Genomic Surveillance Network; GISAID core curation team; Network for Genomic Surveillance in South Africa (NGS-SA); Swiss SARS-CoV-2 Sequencing Consortium; Howden BP, Sintchenko V, Zuckerman NS, Mor O, Blankenship HM, de Oliveira T, Lin RTP, Siqueira MM, Resende PC, Vasconcelos ATR, Spilki FR, Aguiar RS, Alexiev I, Ivanov IN, Philipova I, Carrington CVF, Sahadeo NSD, Branda B, Gurry C, Maurer-Stroh S, Naidoo D, von Eije KJ, Perkins MD, van Kerkhove M, Hill SC, Sabino EC, Pybus OG, Dye C, Bhatt S, Flaxman S, Suchard MA, Grubaugh ND, Baele G, Faria NR. Global disparities in SARS-CoV-2 genomic surveillance. Nature Commun. 2022 Nov 16;13(1):7003. doi: 10.1038/s41467-022-33713-y. PMID: 36385137; PMCID: PMC9667854.
Happi C, Adetifa I, Mbala P, Njouom R, Nakoune E, Happi A, Ndodo N, Ayansola O, Mboowa G, Bedford T, Neher RA, Roemer C, Hodcroft E, Tegally H, O’Toole Á, Rambaut A, Pybus O, Kraemer MUG, Wilkinson E, Isidro J, Borges V, Pinto M, Gomes JP, Freitas L, Resende PC, Lee RTC, Maurer-Stroh S, Baxter C, Lessells R, Ogwell AE, Kebede Y, Tessema SK, de Oliveira T. Urgent need for a non-discriminatory and non-stigmatizing nomenclature for monkeypox virus. PLoS Biol. 2022 Aug 23;20(8):e3001769. doi: 10.1371/journal.pbio.3001769. PMID: 35998195; PMCID: PMC9451062.
Nguyen MN, Krutz NL, Limviphuvadh V, Lopata AL, Gerberick GF, Maurer-Stroh S. AllerCatPro 2.0: a web server for predicting protein allergenicity potential. Nucleic Acids Res. 2022 Jul 5;50(W1):W36-W43. doi: 10.1093/nar/gkac446. PMID: 35640594; PMCID: PMC9252832.
Chong CS, Limviphuvadh V, Maurer-Stroh S. Global spectrum of population-specific common missense variation in cytochrome P450 pharmacogenes. Hum Mutat. 2021 Sep;42(9):1107-1123. doi: 10.1002/humu.24243. Epub 2021 Jun 29.
Konings F, Perkins MD, Kuhn JH, Pallen MJ, Alm EJ, Archer BN, Barakat A, Bedford T, Bhiman JN, Caly L, Carter LL, Cullinane A, de Oliveira T, Druce J, El Masry I, Evans R, Gao GF, Gorbalenya AE, Hamblion E, Herring BL, Hodcroft E, Holmes EC, Kakkar M, Khare S, Koopmans MPG, Korber B, Leite J, MacCannell D, Marklewitz M, Maurer-Stroh S, Rico JAM, Munster VJ, Neher R, Munnink BO, Pavlin BI, Peiris M, Poon L, Pybus O, Rambaut A, Resende P, Subissi L, Thiel V, Tong S, van der Werf S, von Gottberg A, Ziebuhr J, Van Kerkhove MD.SARS-CoV-2 Variants of Interest and Concern naming scheme conducive for global discourse. Nature Microbiol. 2021 Jul;6(7):821-823. doi: 10.1038/s41564-021-00932-w.
Asarnow D, Wang B, Lee WH, Hu Y, Huang CW, Faust B, Ng PML, Ngoh EZX, Bohn M, Bulkley D, Pizzorno A, Ary B, Tan HC, Lee CY, Minhat RA, Terrier O, Soh MK, Teo FJ, Yeap YYC, Seah SGK, Chan CEZ, Connelly E, Young NJ, Maurer-Stroh S, Renia L, Hanson BJ, Rosa-Calatrava M, Manglik A, Cheng Y, Craik CS, Wang CI. Structural insight into SARS-CoV-2 neutralizing antibodies and modulation of syncytia. Cell. 2021 Jun 10;184(12):3192-3204.e16. doi: 10.1016/j.cell.2021.04.033. Epub 2021 Apr 24.
Ooi KH, Liu MM, Tay JWD, Teo SY, Kaewsapsak P, Jin S, Lee CK, Hou J, Maurer-Stroh S, Lin W, Yan B, Yan G, Gao YG, Tan MH. An engineered CRISPR-Cas12a variant and DNA-RNA hybrid guides enable robust and rapid COVID-19 testing. Nature Commun. 2021 Mar 19;12(1):1739. doi: 10.1038/s41467-021-21996-6.
Chua AQ, Al Knawy B, Grant B, Legido-Quigley H, Lee WC, Leung GM, Looi MK, Maurer-Stroh S. How the lessons of previous epidemics helped successful countries fight covid-19. BMJ. 2021 Mar 11;372:n486. doi: 10.1136/bmj.n486.
Young BE, Fong SW, Chan YH, Mak TM, Ang LW, Anderson DE, Lee CY, Amrun SN, Lee B, Goh YS, Su YCF, Wei WE, Kalimuddin S, Chai LYA, Pada S, Tan SY, Sun L, Parthasarathy P, Chen YYC, Barkham T, Lin RTP, Maurer-Stroh S, Leo YS, Wang LF, Renia L, Lee VJ, Smith GJD, Lye DC, Ng LFP. Effects of a major deletion in the SARS-CoV-2 genome on the severity of infection and the inflammatory response: an observational cohort study. Lancet. 2020 Aug 29;396(10251):603-611. doi: 10.1016/S0140-6736(20)31757-8. Epub 2020 Aug 18.
Pung R, Chiew CJ, Young BE, Chin S, Chen MI, Clapham HE, Cook AR, Maurer-Stroh S, Toh MPHS, Poh C, Low M, Lum J, Koh VTJ, Mak TM, Cui L, Lin RVTP, Heng D, Leo YS, Lye DC, Lee VJM; Singapore 2019 Novel Coronavirus Outbreak Research Team. Investigation of three clusters of COVID-19 in Singapore: implications for surveillance and response measures. Lancet. 2020 Mar 28;395(10229):1039-1046. doi: 10.1016/S0140-6736(20)30528-6. Epub 2020 Mar 17
 | MAURER-STROH Sebastian Executive Director, Senior Principal Investigator Email: sebastianms@a-star.edu.sg Research Group: Protein Sequence Analysis |
Dr. Sebastian Maurer-Stroh studied theoretical biochemistry at the University of Vienna and wrote his master and PhD thesis at the Institute of Molecular Pathology (IMP). After FEBS and Marie Curie fellowships at the VIB-SWITCH lab in Brussels, he has been leading a group of experts in protein sequence analysis as a senior principal investigator in the A*STAR Bioinformatics Institute (BII) since 2007. He is Executive Director of BII since January 2021. His protein function analysis skills are supporting A*STAR’s efforts at the public-private interface and through computational analysis and modelling his team is critically contributing to national and global viral pathogen surveillance. He is also adjunct Professor at the National University of Singapore (NUS) and infectious disease expert for A*STAR ID labs and the National Public Health Laboratory of the Ministry of Health, Singapore.
| Principal Scientist | LIMVIPHUVADH Vachiranee |
| Senior Scientist | KENANOV Dimitar |
| Senior Scientist I | MAK Tze Minn Sandy |
| Senior Scientist I | CHIA Jing Ting Charmaine |
| Senior Scientist I | WAILAN Alexander |
| Scientist | CHONG Cheng Shoong Ken |
| Principal Research Officer I | LEE Tze Chuan Raphael |
| Lead Research Officer II | XU Yani Angela |
| Senior Research Officer I | CHEW Yi Hong |
| Senior Research Officer I | NG Ting Ting |
| Research Officer | MAKHEJA Meera |
| PhD Student | ATTIQUE Syed Awais |