
We build computational models that contribute to our understanding of the mechanisms of disease and host immune responses, with a particular focus on infective pathogens. Our main method of choice is molecular simulation, using multiscale approaches that allow us to access a broad range of spatio-temporal scales, from atomically-detailed systems to highly simplified coarse-grained resolutions. We interact closely with our numerous experimental collaborators, and develop new ways to integrate diverse structural, biophysical, and biochemical data into models that enable us to work towards novel therapeutic strategies.
Flaviviruses are enveloped viruses which encompass numerous human pathogens of escalating global health concern that are predominantly transmitted by mosquitoes and ticks. Traditionally, we have focused on dengue virus, which represents a significant burden to human health and the economy in Singapore. We have developed “virtual flavivirus” models that have been used to elucidate key viral dynamics associated with infection and antibody recognition [1]. As part of a collaborative team across Duke-NUS and A*STAR funded by an NRF CRP grant, we are pursuing mRNA-based vaccine development to target all four dengue serotypes. In parallel, we are exploring other vaccine technologies, including engineering of dengue virus-like particles (VLPs) [Patent No. 10202303148T; 6th November 2023] supported by a YIRG grant awarded to Jan K. Marzinek (Figure 1A). Nevertheless, dengue exhibits a phenomenon known as antibody-dependent enhancement which can exacerbate infections and complicates traditional vaccine-based approaches. As an alternative approach, the pH-dependent conformational changes of the envelope protein required for fusion are attractive points for inhibition. To this end, we have developed a computational platform to probe membrane proteins for hidden druggable pockets, which has been used to uncover a novel cryptic site in flaviviruses associated with pH-sensing and is now being pursued for potential antivirals [2].
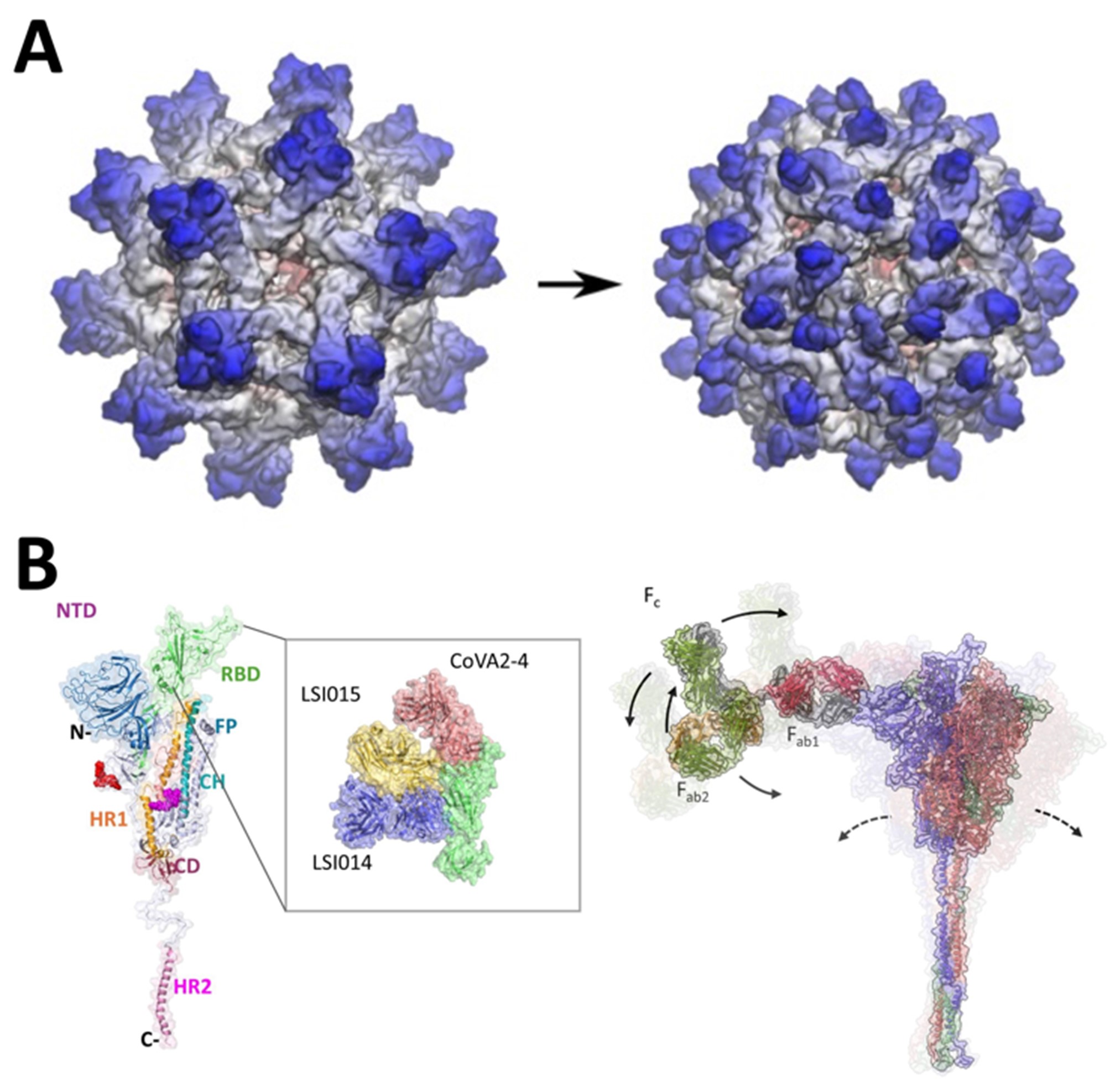
Figure 1. Viral therapeutics. (A) Predicting the maturation process of dengue VLPs, towards improved secretion properties for use in vaccines. (B) SARS-CoV-2 spike protein modelling for (left) mapping receptor binding domain (RBD) epitopes of novel antibodies, and (right) predicting allosteric motions induced by full-length IgG antibody.
We are leveraging the methods we have developed for flaviviruses to other enveloped viruses including SARS‑CoV‑2. Supported by an ID HTPO Seed Fund grant, we have characterized the structure and dynamics of the viral surface spike protein for a wide range of SARS-CoV-2 variants. We computationally mapped its glycan coating as part of a global collaboration [3], showing that constructs derived from different labs are highly similar and closely resemble their viral counterpart, helping to rationalize the success of spike-based vaccines. A multidisciplinary approach has also revealed that the spike protein plays previously undefined pathogenic roles in serious COVID-19 infections [4, 5] including augmenting hyperinflammation by acting as a conduit to over-activate host immune pathways. Our findings highlight potential therapeutic strategies for future viral infections that cause over-amplified immune reactions and sepsis. Furthermore, we have discovered the importance of long-range allostery [1] in governing spike function [6]. We showed with collaborators at NUS that antibodies may exploit such allostery to tune neutralizing efficacy, while also exhibiting synergistic binding behavior which may offer potential routes to antiviral “cocktail” development (Figure 1B). Finally, we have leveraged our platform for probing membrane protein pockets [2] to uncover potentially druggable sites on the spike protein [8]; looking towards future epidemic preparedness, we are aiming to develop fusion-inhibitory therapeutics by targeting conserved sites across different coronaviruses, while also bearing in mind the potential for such viruses to evolve alternative mechanisms of infection [9].
Antimicrobial resistance (AMR) is a leading cause of death worldwide, and there is an urgent need to discover new ways to battle AMR infections. For example, as part of a collaboration with P&G, we are establishing computational approaches to predict the modes of action of disinfectants against viral capsids and are working with collaborators at the Universities of Southampton and Oxford to better understand the impact of hand sanitizer ingredients upon pathogen envelopes [10]. A multiscale simulation approach can provide an improved understanding of bacterial membrane structure and function [11-14] and contribute towards the development of novel strategies to battle AMR [15]. For example, in collaboration with NTU, we have helped to clarify the mechanisms of action of drugs that inhibit the essential F-ATP synthase enzyme in mycobacteria, which cause diseases such as tuberculosis, and are leveraging this to target other AMR species [16-18]. We are also interested in developing strategies to target the excessive activation of inflammatory pathways that can occur during systemic infections by AMR bacteria. In a long-running collaboration with researchers at Lund University, a cyclic molecule has been developed based on the evolutionarily conserved innate structural fold of endogenous thrombin-derived C-terminal peptides (TCPs) which targets both bacterial toxins and host immune receptors [19] (Figure 2A). We have also helped to show how such TCPs may be incorporated into sutures to reduce the incidence of surgical site infections [20] (Figure 2B).
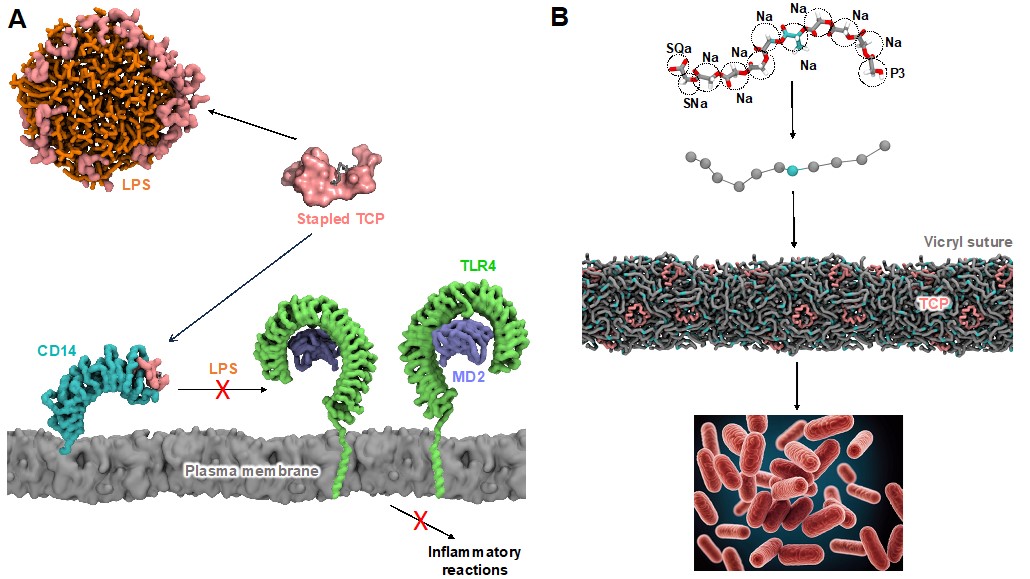
Figure 2.: Exploiting host defence peptides for therapeutics. (A) A hydrophobic staple was added to TCP to develop a dual-action drug that both neutralizes bacterial lipopolysaccharide (LPS) and inhibits the host CD14/TLR4 pathway to block inflammatory reactions. (B) Multiscale models of Vicryl suture polymers were used to predict the stable incorporation of TCPs, a strategy which was then demonstrated to successfully prevent surgical site infections.
We have a broad interest in other challenges within biophysics [21], bionanotechnology [22], and biomedicine [23]. For example, as part of an NMRC OF-IRG grant with Duke-NUS and NTU, we are focused on overcoming drug resistance in cancer due to ABCB1 transporter overexpression, and recently reported a novel cyclic tryptophan-based molecule which inhibits ABCB1 with an efficacy better than classical inhibitors [23]. We also contribute important molecular simulation methodological developments, such as new approaches to calculate protein glass transition temperatures which correlate with their biochemical activities and may be modulated by bioprotective compounds [24], and coarse-grained forcefields for describing the glycosaminoglycans (GAGs) in proteoglycans which play vital roles in numerous physiological and pathological processes, particularly governing cellular communication and cell surface attachment [25] (Figure 3).
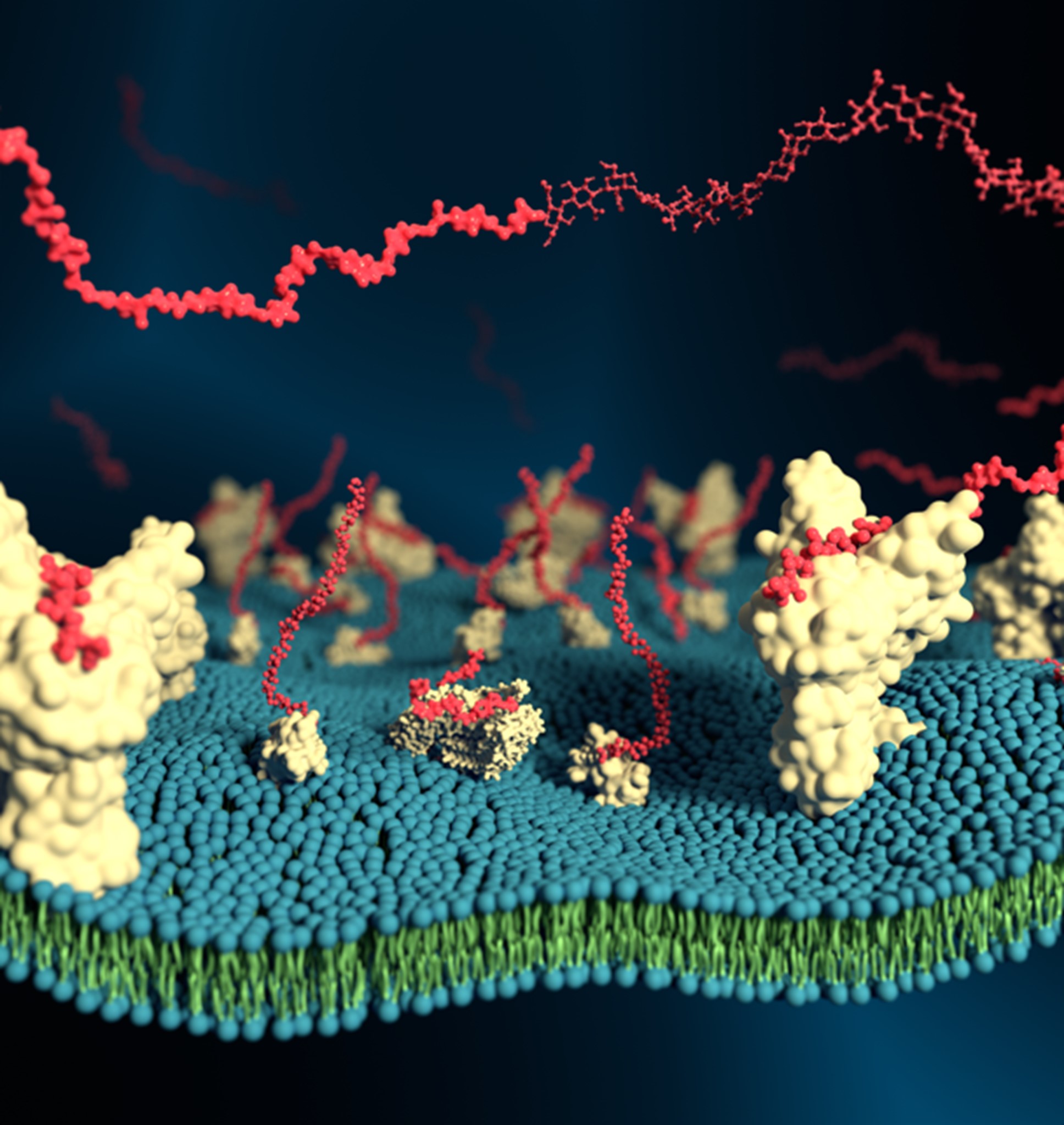
Figure 3. Multiscale modelling of GAGs. Physiologically realistic mammalian cell surfaces may be modelled by simulating GAGs (red) on membrane-bound proteoglycans and receptors.
| Senior Principal Investigator | BOND Peter J. | [View Bio] |
| Senior Scientist | MARZINEK Jan |
| Senior Scientist | SAMSUDIN Mohd Firdaus |
| Scientist | PALUR Venkata Raghuvamsi |
| Scientist | SOMBOON Kamolrat |
| Scientist | MASIREVIC Srdan |
| PhD Students | HAREED Ayub Mohamed |
Samsudin F, Zuzic L, Marzinek JK, Bond PJ. (2024). Mechanisms of allostery at the viral surface through the eyes of molecular simulation. Curr Opin Struct Biol. 84:102761.
Zuzic L, Marzinek JK, Anand GS, Warwicker J, Bond PJ. (2023). A pH-dependent cluster of charges in a conserved cryptic pocket on flaviviral envelopes. Elife. 12:e82447.
Chawla H, Jossi SE, Faustini SE, Samsudin F, Allen JD, Watanabe Y, Newby ML, Marcial-Juárez E, Lamerton RE, McLellan JS, Bond PJ, Richter AG, Cunningham AF, Crispin M. (2022). Glycosylation and Serological Reactivity of an Expression-enhanced SARS-CoV-2 Viral Spike Mimetic. J Mol Biol. 434:167332.
Petrlova J, Samsudin F, Bond PJ, Schmidtchen A. (2022). SARS-CoV-2 spike protein aggregation is triggered by bacterial lipopolysaccharide. FEBS Lett. 596:2566-2575.
Samsudin F, Raghuvamsi P, Petruk G, Puthia M, Petrlova J, MacAry P, Anand GS, Bond PJ*, Schmidtchen A*. (2023) SARS-CoV-2 spike protein as a bacterial lipopolysaccharide delivery system in an overzealous inflammatory cascade. J Mol Cell Biol. 14:mjac058. (Journal Cover Article).
Tan ZW, Tee WV, Samsudin F, Guarnera E, Bond PJ, Berezovsky IN. (2022). Allosteric perspective on the mutability and druggability of the SARS-CoV-2 Spike protein. Structure. 30:590-607.e4.
Tulsian NK, Raghuvamsi PV, Qian X, Yue G, Shunmuganathan BD, Samsudin F, Hwa WY, Jianqing L, Purushotorman K, Kozma MM, Wang B, Lescar J, Wang CI, Gupta RK, Bond PJ*, MacAry PA*. (2023). Defining neutralization and allostery by antibodies against COVID-19 variants. Nature Commun. 14:6967.
Zuzic L, Samsudin F, Shivgan AT, Raghuvamsi PV, Marzinek JK, Boags A, Pedebos C, Tulsian NK, Warwicker J, MacAry P, Crispin M, Khalid S, Anand GS, Bond PJ. (2022). Uncovering cryptic pockets in the SARS-CoV-2 spike glycoprotein. Structure. 30:1062-1074.e4.
Mah MG, Linster M, Low DHW, Yan Z, Jayakumar J, Samsudin F, Wong FY, Bond PJ, Mendenhall IH, Su YCF, Smith GJD. (2023). Spike-independent infection of human coronavirus 229E in bat cells. Microbiol. Spectr. 11:e0348322.
Waller C, Marzinek JK, McBurnie E, Bond PJ*, Williamson PTF*, Khalid S*. (2023). Impact on S. aureus and E. coli membranes of treatment with chlorhexidine and alcohol solutions: insights from molecular simulations and nuclear magnetic resonance. J Mol Biol. 435:167953.
Khalid S, Schroeder C, Bond PJ, Duncan AL. (2022). What have molecular simulations contributed to understanding of Gram-negative bacterial cell envelopes? Microbiology. 168:001165.
Guilvout I, Samsudin F, Huber H, Bond PJ, Bardiaux B, Francetic O. (2024). Membrane platform protein PulF of the Klebsiella type II secretion system forms a trimeric ion channel essential for endopilus assembly and protein secretion. mBio. 15:e0142323.
Tupiņa D, Krah A, Marzinek JK, Zuzic L, Moverley AA, Constantinidou C, Bond PJ. (2022). Bridging the N-terminal and middle domains in FliG of the flagellar rotor. Curr Res Struct Biol. 4:59-67.
Krah A, Vogelaar T, de Jong SI, Claridge JK, Bond PJ, McMillan DGG. (2023). ATP binding by an F1Fo ATP synthase ε subunit is pH dependent, suggesting a diversity of ε subunit functional regulation in bacteria. Front Mol Biosci. 10:1059673.
Puthia M, Marzinek JK, Petruk G, Ertürk Bergdahl G, Bond PJ, Petrlova J. (2022). Antibacterial and Anti-Inflammatory Effects of Apolipoprotein E. Biomedicines. 10:1430.
Ragunathan P, Sae-Lao P, Hamela C, Alcaraz M, Krah A, Poh WH, Ern Pee CJ, Hou Lim AY, Rice SA, Pethe K, Bond PJ, Dick T, Kremer L, Bates RW, Grüber G. (2024). High efficacy of the F-ATP synthase inhibitor TBAJ-5307 against nontuberculous mycobacteria in vitro and in vivo. J Biol Chem. 300:105618.
Krah A, Ragunathan P, Bond PJ, Grüber G. (2024). Variations of the Mycobacterium abscessus F-ATP synthase subunit a-c interface alter binding and potency of the anti-TB drug bedaquiline. Biochem Biophys Res Commun. 690:149249.
Krah A, Grüber G, Bond PJ. (2022). Binding properties of the anti-TB drugs bedaquiline and TBAJ-876 to a mycobacterial F-ATP synthase. Curr Res Struct Biol. 4:278-284.
Petruk G, Puthia M, Samsudin F, Petrlova J, Olm F, Mittendorfer M, Hyllén S, Edström D, Strömdahl AC, Diehl C, Ekström S, Walse B, Kjellström S, Bond PJ, Lindstedt S, Schmidtchen A. (2023). Targeting Toll-like receptor-driven systemic inflammation by engineering an innate structural fold into drugs. Nature Commun. 14:6097.
Puthia M, Petrlova J, Petruk G, Butrym M, Samsudin F, Andersson MA, Strömdahl AC, Wasserstrom S, Hartman E, Kjellström S, Caselli L, Klementieva O, Bond PJ, Malmsten M, Raina DB, Schmidtchen A. (2023). Bioactive suture with added innate defense functionality for the reduction of bacterial infection and inflammation. Adv Healthc Mater. e2300987. (Journal Cover Article).
Kolloff C, Mazur A, Marzinek JK, Bond PJ, Olsson S, Hiller S. (2022). Motional clustering in supra-τc conformational exchange influences NOE cross-relaxation rate. J Magn Reson. 338:107196.
Dutta C, Krishnamurthy P, Su D, Yoo SH, Collie GW, Pasco M, Marzinek JK, Bond PJ, Verma C, Grélard A, Loquet A, Li J, Luo M, Barboiu M, Guichard G, Kini RM, Kumar PP. (2023). Nature-inspired synthetic oligourea foldamer channels allow water transport with high salt rejection. Chem. 9:2237-2254.
Koh J, Itahana Y, Krah A, Mostafa H, Ong MM, Iwamura S, Vincent DM, Krishnan SR, Ye W, PWC Yim, Khopade TM, Chen K, Kong PS, Wang LF, Bates RW, Kimura Y, Viswanathan R*, Bond PJ*, Koji Itahana*. (2024). From Bats to Drugs: Exploring Cyclic Tryptophan Diketopiperazine as ABCB1 Inhibitors. Commun Chem. In press.
Li E, Yaochai MY, Ng JM, Huber RG, Bond PJ, Krah A. (2023). Observation of multiple protein temperature transitions dependent upon the chemical environment. J Mol Liq. 385:122348.
Shivgan AT, Marzinek JK, Krah A, Matsudaira P, Verma CS, Bond PJ. (2024). Coarse-Grained Model of Glycosaminoglycans for Biomolecular Simulations. J Chem Theory Comput. 20:3308-3321. (Journal Cover Article).
 | BOND Peter J. Senior Principal Investigator Email: peterjb@a-star.edu.sg Research Group: Multiscale Simulation, Modelling and Design |
Peter J. Bond is Senior Principal Scientist of the Multiscale Simulation, Modeling and Design (MSMD) group at the Bioinformatics Institute (BII) A*STAR, Singapore, and Adjunct Associate Professor at the National University of Singapore (NUS). His research interests include host-pathogen interactions, membrane receptor signaling, virus dynamics, and therapeutics development. Following his graduation in Biochemistry at the University of Oxford in 2001, Peter moved to the Laboratory of Molecular Biophysics to read for a DPhil, supported by a Wellcome Trust Prize Studentship, and was subsequently awarded an EMBO Long-Term Fellowship hosted at the Max Planck Institute of Biophysics in Frankfurt. In 2010, he became a University Lecturer at the University of Cambridge, before moving to Singapore in late 2013.
| Senior Scientist | MARZINEK Jan |
| Senior Scientist | SAMSUDIN Mohd Firdaus |
| Scientist | PALUR Venkata Raghuvamsi |
| Scientist | SOMBOON Kamolrat |
| Scientist | MASIREVIC Srdan |
| PhD Students | HAREED Ayub Mohamed |