
Coronary artery disease (CAD) affects 6% of the general population and up to 20% of those over 65. It is a leading cause of cardiac mortality, responsible for 19% of deaths in Singapore (MOH website). The incidence of CAD is increasing due to aging and higher diabetes prevalence.
Computed Tomography Coronary Angiography (CTCA) is the recommended first-line investigation for CAD, as per updated NICE guidelines. Studies like PROMISE and SCOT-HEART support CTCA for evaluating coronary anatomy and physiology, showing it enhances diagnostic certainty, improves triage efficiency to invasive catheterization, and reduces radiation exposure compared to functional stress testing.
Currently, CAD report generation requires 1-3 hours of a CT specialist’s time with a 20% inter-observer variability. There is also a lack of effective tools to analyze Agatston scores, stenosis severity, and plaque characterization, limiting CTCA’s diagnostic and research utility.
Leveraging Singapore’s strengths in artificial intelligence (AI), we aim to develop an AI-driven CT Coronary Angiography platform. This platform will automate anonymization, reporting, Agatston scoring, and plaque quantification. It will serve as a comprehensive solution from diagnosis to clinical management, prognosis, and therapy response prediction for the pharmaceutical industry.
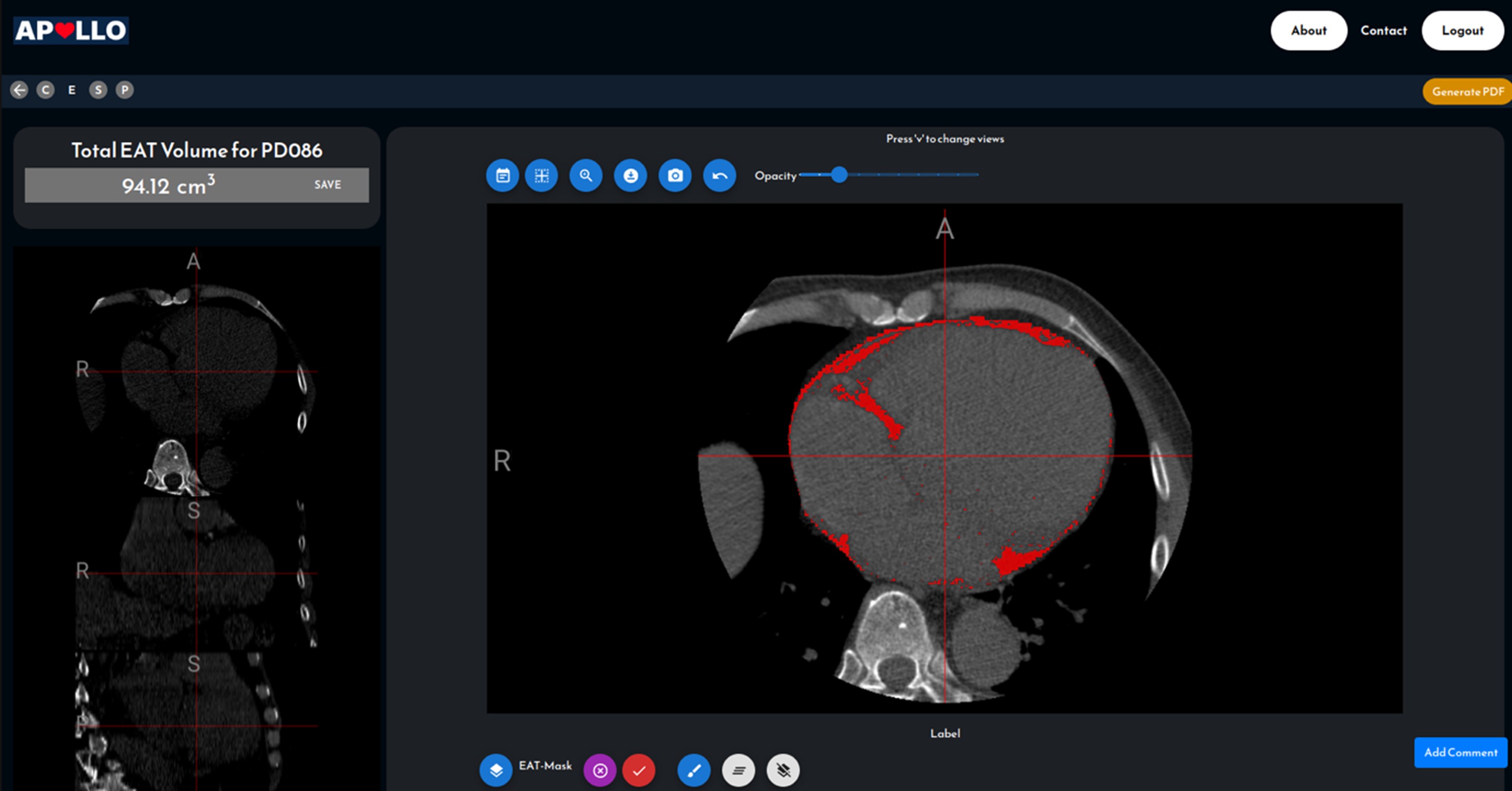
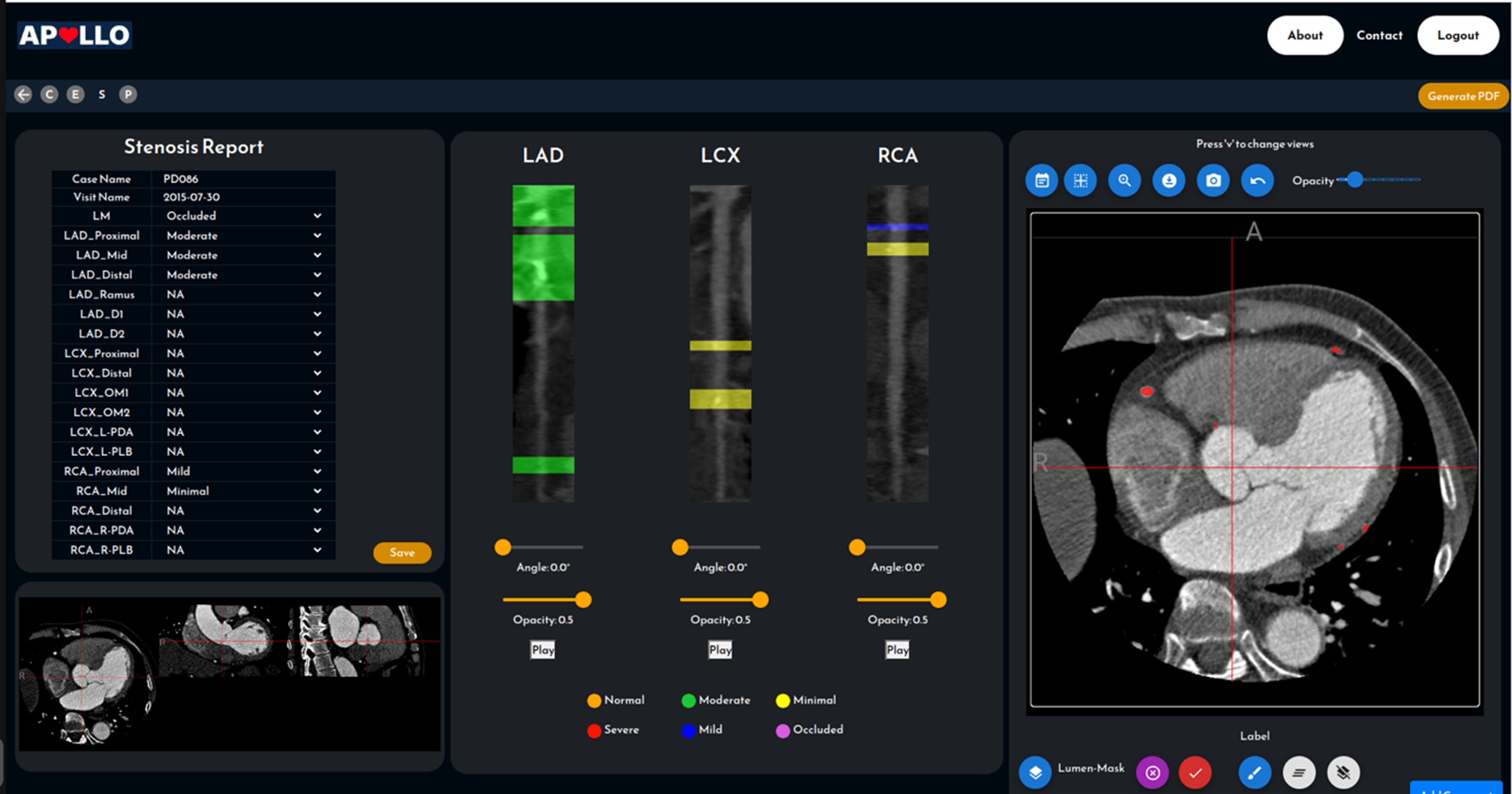
Figure 1.
We work on spatial omics research in collaborative with Genome Institute of Singapore, Singapore Immunology Network, National Cancer Centre Singapore, Singapore General Hospital and Duke-NUS Medical School. By utilizing spatial omics techniques, such as MERFISH, we can detect the expression and physical location of thousands of DNA and RNA molecules. This approach allows us to study the spatial organization, cell types, and intercellular communication. Specifically, we proposed a new method called BANKSY1, which achieves more accurate cell type clustering by augmenting the transcriptomic profile of each cell with a weighted sum of its spatial neighbors' transcriptomes. Additionally, we investigated DNA copy number variation (CNV) in colorectal cancer and have identified distinct CNV profiles of colorectal subtypes, which can aid in cancer state prediction and prognostic treatment.
Related Publication
Singhal, V., Chou, N., Lee, J., Yue, Y., Liu, J., Chock, W. K., ... & Prabhakar, S. (2024). BANKSY unifies cell typing and tissue domain segmentation for scalable spatial omics data analysis. Nature Genetics, 1-11.
As part of a collaborative effort with the Institute of Materials Research and Engineering ASTAR (IMRE) we developed a research support application to monitor the growth of plants during the germination stage. The application uses computer vision to detect the plants from mobile phone images and can compute useful metrics regarding their growth. The application also includes an easy-to-use graphical interface that allows the user to set up the experiments, import and analyze the images.
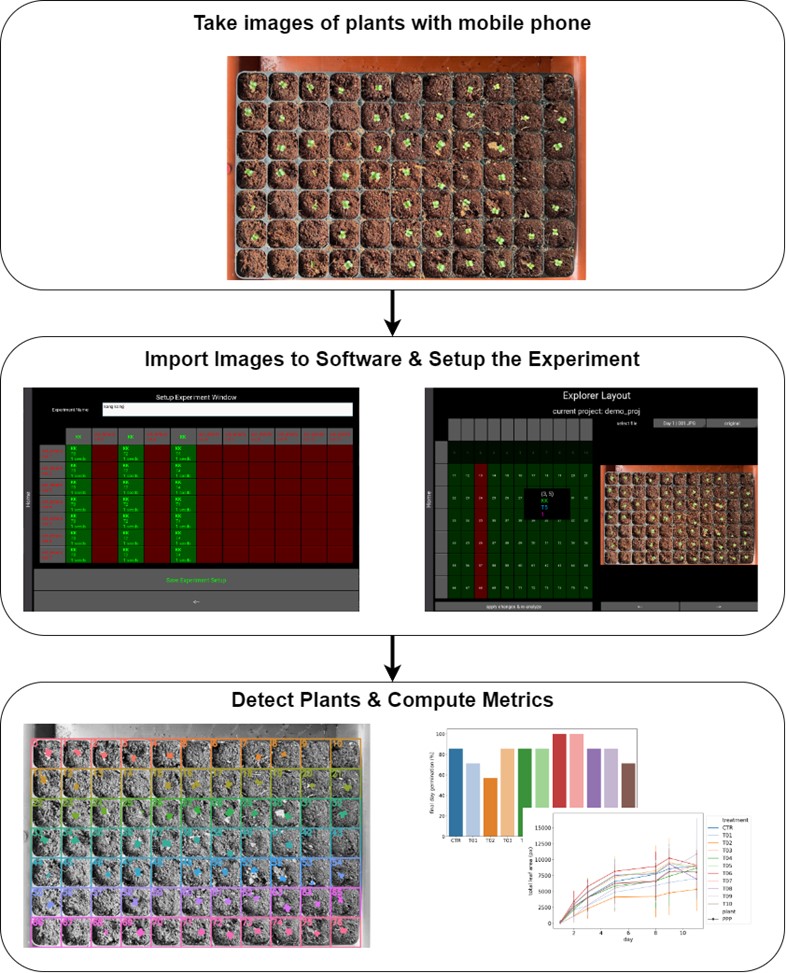
Figure 2. processing pipeline of plant monitoring software
Coronary angiography is the gold standard imaging technique in the diagnosis of coronary artery disease. This method involves the acquisition of moving X-ray images from various angles around the patient's torso following the injection of iodine-based contrast. Traditionally, the analysis and interpretation of coronary angiogram is performed by trained cardiologists. However, the process can be time-consuming and subject to inter-observer variability. In collaboration with the National Heart Centre Singapore, our project explores AI approaches to automate the analysis of angiogram video sequences, aiming to develop a quantitative assessment tool that provides repeatable and objective measurements.
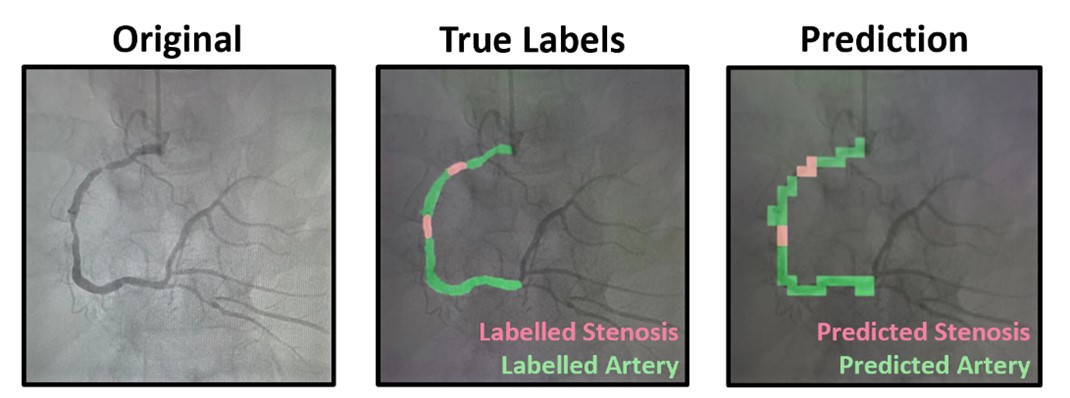
Figure 3. In the left panel, the original coronary angiography frame is displayed. The middle panel illustrates the annotated coronary artery (highlighted in green) and stenosis (highlighted in red) on the coronary angiography frame. The right panel shows the model's prediction of the coronary artery (highlighted in green) and stenosis (highlighted in red).
Cancer is a leading global cause of death, responsible for 9.7 million deaths in 2022. Cancer begins with cells acquiring mutations, growing uncontrollably, and forming tumors. With latest advances in precision medicine, clinicians are using molecular level information to design treatment. For example, tumor purity (fraction of cancer cells out of all cells in given tissue area), FGA (Fraction Genome Altered), EGFR mutation status, etc, are needed for better cancer treatment. These molecular level information needs additional resources in the current clinical workflow.
Currently pathologists review the whole slide images as the first assessment of cancer. The (expensive) molecular tests are carried out as and when needed and are not in routine workflow. Our team is developing deep learning models to quantify tumor by analyzing histopathology images. One such model, based on Multiple Instance Learning (MIL), predicts tumor purity from digital slides. Accurate tumor purity estimation is crucial for selecting samples for genomic sequencing. Our tumor purity model has shown high consistency with genomic tumor purity values across eight TCGA cohorts and a local Singapore cohort. Our model also provides tumor purity maps, revealing spatial variation within sections and aiding in understanding the tumor microenvironment.
Additionally, we are developing models for tumor staging, mutation analysis, and survival analysis. Our aim is to support medical professionals in diagnosis, treatment planning, medication management, and precision medicine, addressing future healthcare demands more effectively.
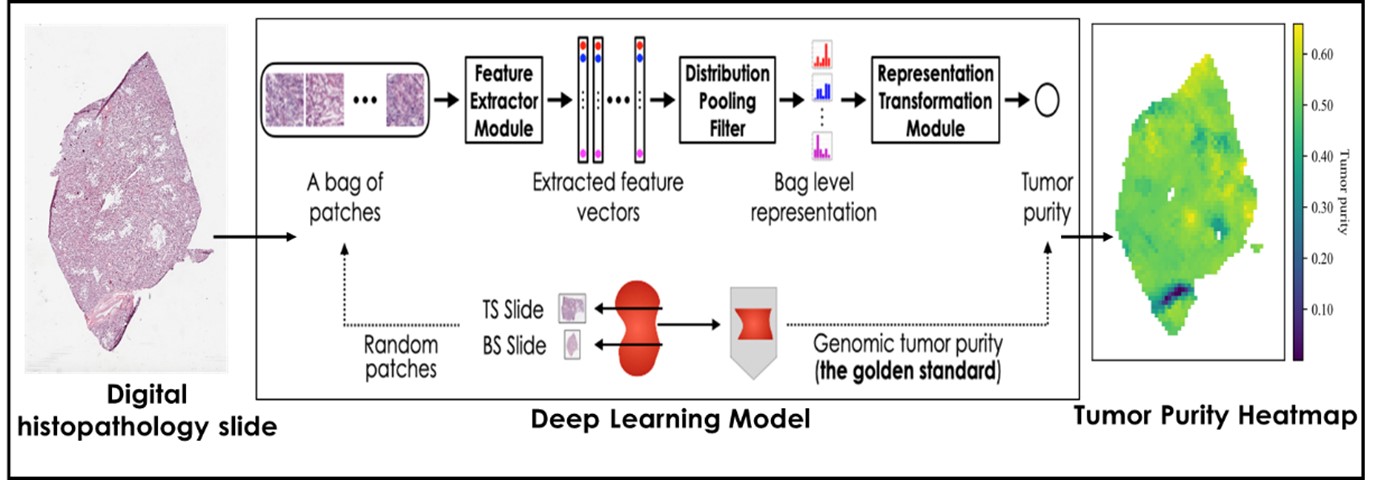
Figure 4. Spatial Tumor Purity Maps [1] facilitates:
Related Publication
Oner, M. U., Chen, J., Revkov, E., James, A., Heng, S. Y., Kaya, A. N., ... & Lee, H. K. (2022). Obtaining spatially resolved tumor purity maps using deep multiple instance learning in a pan-cancer study. Patterns, 3(2)
Identifying disease targets remains a major challenge due to the lack of access to high-quality molecular data at the "scene of the crime", i.e., within the context of the diseased tissue. Spatial omics (SO) technology has emerged as a powerful tool for high-throughput profiling of RNAs within tissues. However, SO data is expensive and difficult to collect, and has not yet been proven in the clinic. Conversely, histopathological whole-slide imaging (WSI) is routinely used in histopathology for diagnosis and prognosis, yet it lacks the molecular insights crucial for biomarker discovery.
This project pioneers a tri-modal AI framework that integrates molecular (SO), histopathological (WSI), and clinical data to identify novel disease targets. By leveraging national-scale data repositories, including an extensive Asian disease cohort, our integrative model unlocks the latent potential of existing WSI and clinical data resources through multi-modal learning. A central aim of this effort is the development of an SO data generator that can impute spatial omics profiles from WSI data. This generator will address the roadblock of SO data scarcity and pave the way for the discovery of novel targets.
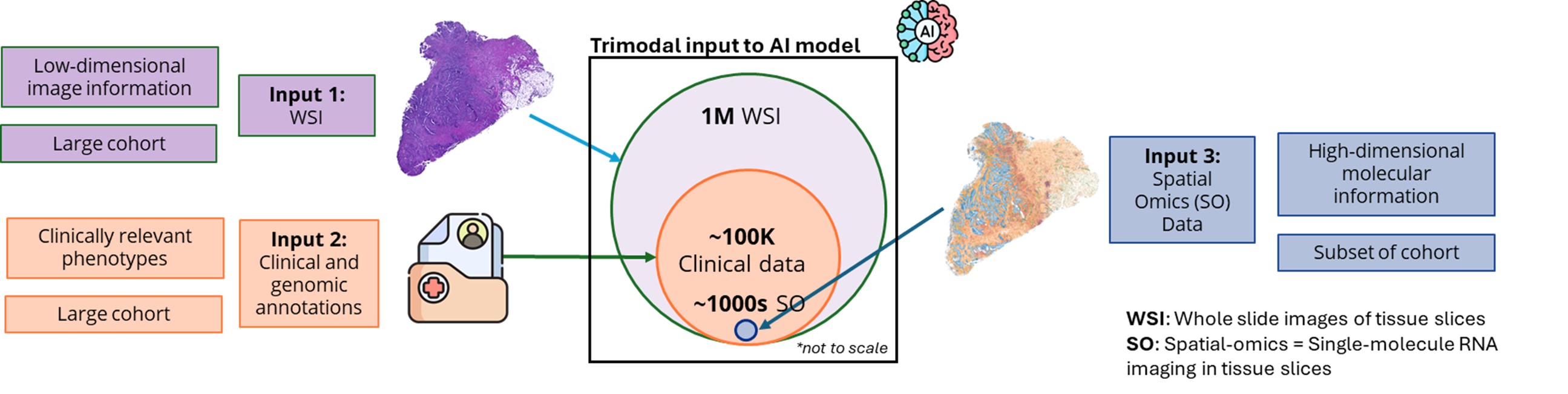
Figure 5. Trimodal AI Framework for RNA Target Discovery
| Deputy Director (Training and Talent) / Senior Principal Scientist | LEE Hwee Kuan | [View Bio] |
| Senior Scientist | LIU Wei |
| Senior Scientist | SINGH Malay |
| Post Doc Research Fellow (Collaborator) | PARK Sojeong |
| Lead Research Officer (T-UP) | SRINIVASA Channarayapatna Arvind |
| Senior Research Officer | GOH Jie Hui Corinna |
| Research Officer | ZHANG Tianyi |
| Research Officer | CHOO Yu Liang |
| Research Officer | SHIMAZAWA Kei |
| Research Officer | HESTER James |
| PhD Student | REN Yu |
 | LEE Hwee Kuan (Head) Deputy Director (Training and Talent) / Senior Principal Scientist Email: leehk@a-star.edu.sg Research Group: Computer Vision and Pattern Discovery |
Dr. Lee Hwee Kuan is a Senior Principal Investigator of the Imaging Informatics division in Bioinformatics Institute. His current research work involves developing of computer vision aglorithms for clinical and biological studies. Dr. Lee obtained his Ph.D. in 2001 in Theoretical Physics from Carnegie Mellon University with a thesis on liquid-liquid phase transitions and quasicrystals. He then held a joint postdoctoral position with Oak Ridge National Laboratory (USA) and University of Georgia where he worked on developing advanced Monte Carlo methods and nano-magnetism. In 2003, with an award from the Japan Society for Promotion of Science, Hwee Kuan moved to Tokyo Metropolitan University where he developed solutions to extremely long time scaled problems and a reweighting method for nonequilibrium systems. In 2005, Dr. Lee returned to Singapore and join Data Storage Institute, A*STAR on investigating novel recording methods such as hard disk recording via magnetic resonance. In 2006, Dr. Lee transferred to BII, A*STAR as Principal Investigator and he leaded the Imaging Informatics Division.
| Senior Scientist | LIU Wei |
| Senior Scientist | SINGH Malay |
| Post Doc Research Fellow (Collaborator) | PARK Sojeong |
| Lead Research Officer (T-UP) | SRINIVASA Channarayapatna Arvind |
| Senior Research Officer | GOH Jie Hui Corinna |
| Research Officer | ZHANG Tianyi |
| Research Officer | CHOO Yu Liang |
| Research Officer | SHIMAZAWA Kei |
| Research Officer | HESTER James |
| PhD Student | REN Yu |