
Our group works at the interface of chemistry and biology. We complement experimental efforts in chemical biology and drug discovery by using computational tools to understand how chemicals interact with biological systems. A central tenet of our research is the use of concepts from fragment-based drug discovery.
The identification of binding pockets is an essential component of the drug discovery process. We have developed a computational pocket detection method that combines ideas from fragment-based drug discovery with molecular dynamics (MD) simulations to account for the effects of protein flexibility and solvation. In this method, which we call ligand-mapping molecular dynamics (LMMD), small organic molecules called fragments are incorporated into the protein’s solvent box to probe for binding pockets during MD simulations. LMMD was first used with benzene probes to design a ligand to target a recalcitrant cryptic pocket (Tan et al., 2012). The method has since been employed in various studies to interrogate a range of proteins, providing crucial insights that have led to the identification of druggable cryptic pockets and discovery of novel protein–protein interaction inhibitors, thus underlining its usefulness for drug discovery.
Through the incorporation of multiple types of fragment probes with different functional groups, LMMD has been extended to the simultaneous identification of hydrophobic, polar, charged, and cryptic binding sites (Tan and Verma, 2020). We have recently developed a new implementation of the method called accelerated LMMD (aLMMD), which is used to detect highly elusive binding sites (Figure 1). These binding sites include recalcitrant cryptic pockets, which are deeply buried pockets that require extensive conformational movement to expose, and occluded binding sites that are buried beneath the protein surface and have no access to the solvent. This method is expected to be a valuable tool for structure-based drug discovery as it can be used to create a comprehensive profile of ligand binding sites in protein targets. We are currently developing an arsenal of LMMD-based approaches, which are expected to provide new insights into the structure and dynamics of drug targets, and guide the design of therapeutics and chemical tools for biological applications. Parallel to these developments, we are also developing tools that combine artificial intelligence (AI) with fragment-based principles to design new drug molecules. We are keen to apply our methods to chemical biology and drug discovery projects and welcome collaborations with groups working in these areas.
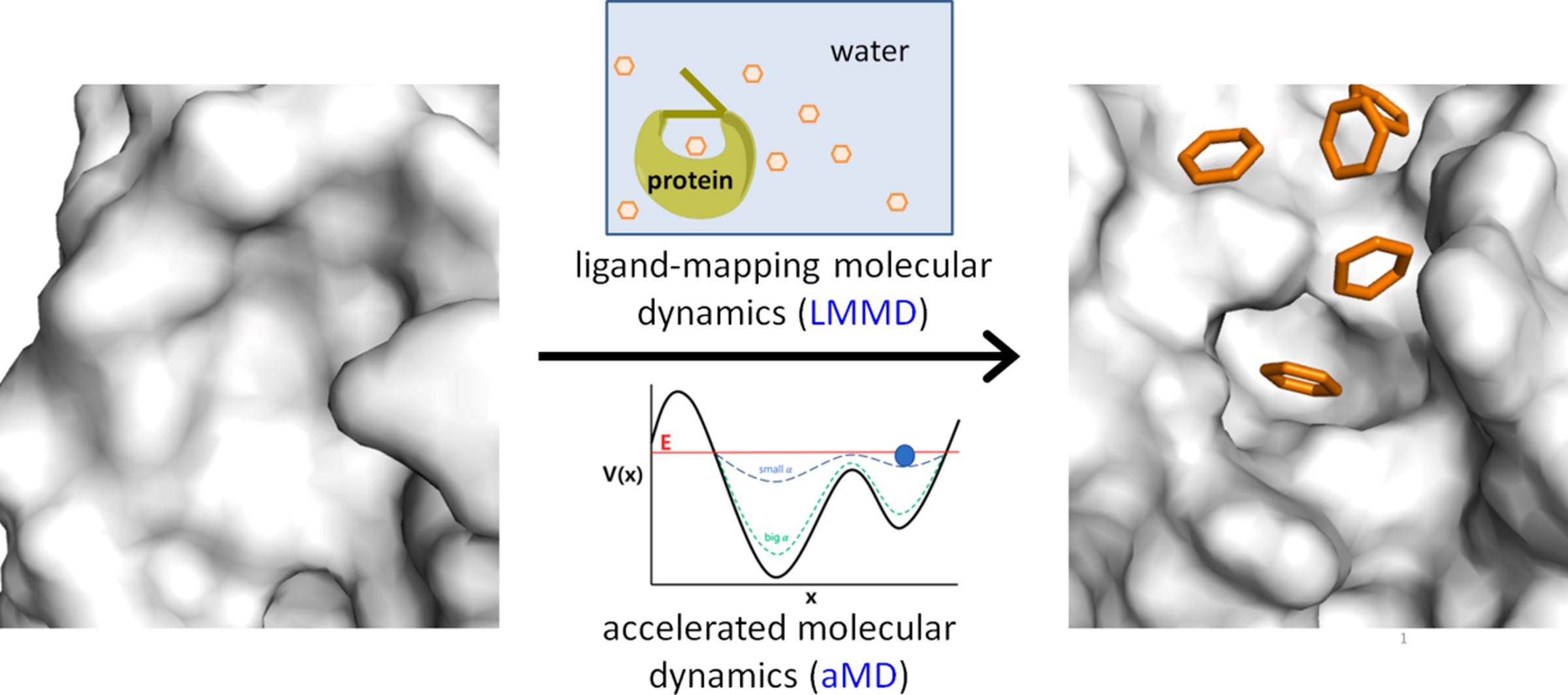
Figure 1. Accelerated LMMD combines LMMD and aMD to reveal recalcitrant cryptic binding pockets (J. Chem. Theory Comput. 2022).
Structure-based design of next-generation therapeutics
Aptamers are single-stranded DNA or RNA molecules with unique tertiary structures that enable them to bind to a variety of target molecules with high affinity and specificity. They hold great potential as therapeutics because of their high binding specificity and low immunogenicity. So far, one aptamer has been approved for therapeutic application while several others have advanced to clinical trials. We are currently working with researchers in A*STAR to apply structure-based computational approaches, such as LMMD and docking, to the rational design of therapeutic aptamers.
Furthermore, we are involved in the structure-based design of a certain class of therapeutics called constrained or stapled peptides. The formation of a covalent “staple” between two appropriately positioned amino acid residues in the peptide constrains its conformation, which helps to enhance binding potency, protease resistance, and to a certain degree, cell permeability. Our computational modelling efforts have led to the development of the first bioactive stapled non-helical peptides with collaborators in the University of Cambridge. These studies show that peptides can also be stabilised in extended conformations by stapling, thus opening up a whole new spectrum of protein–protein interactions for therapeutic targeting.
Understanding biomolecular mechanisms
We collaborate closely with IMCB to study the molecular mechanisms of diabetes-causing mutations. We have specifically studied mutations that cause two forms of monogenic diabetes, neonatal diabetes mellitus and maturity onset diabetes of the young (MODY). These mutations occur in insulin and HNF1A respectively. Through the use of MD simulations, the detrimental effect of the mutations on protein structure and function were elucidated. We have recently obtained a grant to extend our study to type 2 diabetes (T2D)-linked coding variants, which will hopefully help to shed light on the complex molecular pathogenesis of T2D, identify new drug targets for personalised therapy, and aid the development of precision medicine approaches with improved clinical outcomes.
We also collaborate with biologists in IMCB to provide insights into the molecular mechanisms of biological processes.
Recently, our work provided insight into the molecular mechanism of transcriptional repression by DNA topoisomerase 1 (TOP1) (Figure 2). This helped to guide the design of experiments to validate the interaction between TOP1 and DNA and explain the functional consequences of mutations that occur at the secondary DNA binding interface of TOP1.
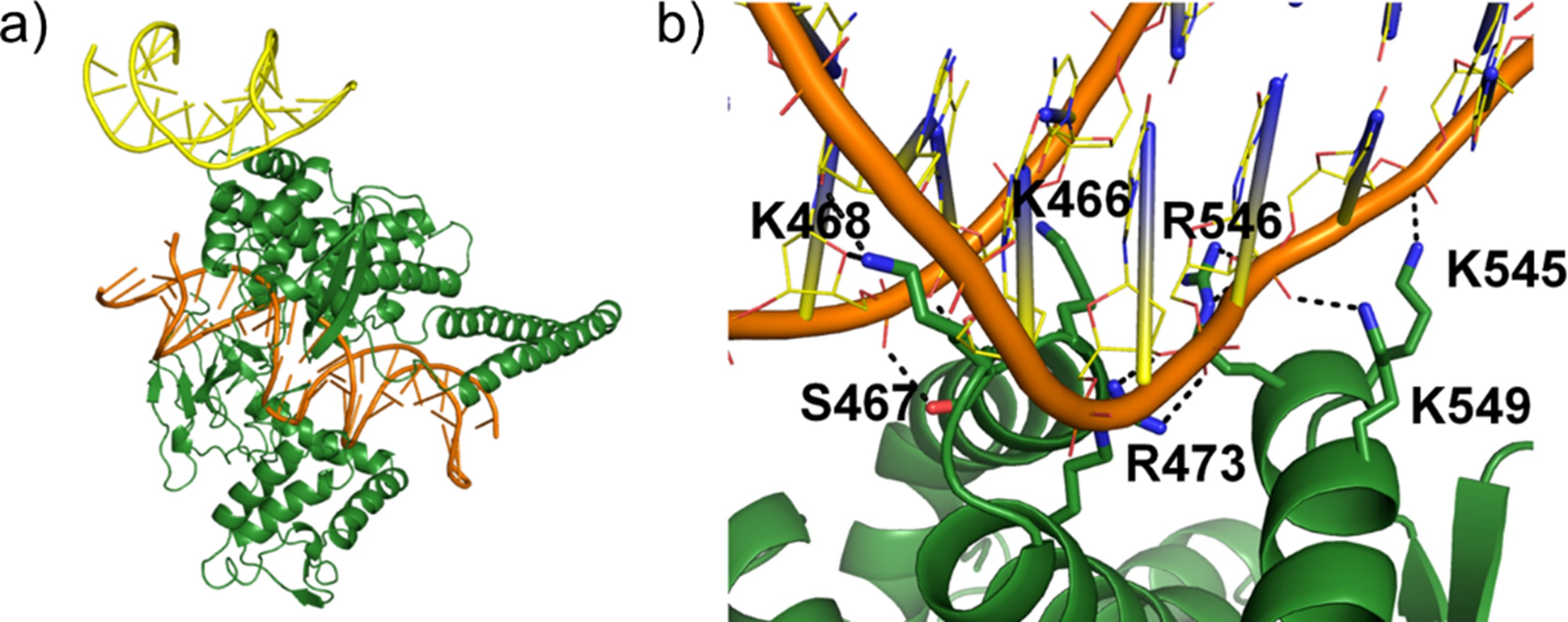
Figure 2. Molecular mechanism of transcriptional repression by a secondary DNA binding interface of TOP1. (a) Molecular model of human TOP1 covalent complex (TOP1 in green, DNA in orange) in complex with DNA at secondary binding site (yellow). (b) MD simulations predicted novel secondary interface residues that have not been reported before. A mutation to one of these residues (R546Q) was shown to destabilise DNA binding. (Nat. Commun. 2023)
Bioelectronics
The group also has a productive collaboration with SUTD in the field of bioelectronics, which marries biology and electronics for biomedical applications. We use molecular modelling techniques to reveal the mechanisms of bioelectronic sensors developed by our collaborator, Prof. Desmond Loke, and suggest modifications to improve the performance of these devices (Figure 3). Biosensors have been developed to detect human embryonic stem cells and differentiate cancer cells from healthy cells, and a new electroporation programming scheme was developed to increase membrane recovery time. Most recently, we have combined two-dimensional (2D) materials with viral particles for electrothermal therapy and tumour ablation, which paves the way for the use of 2D materials in cancer therapy.
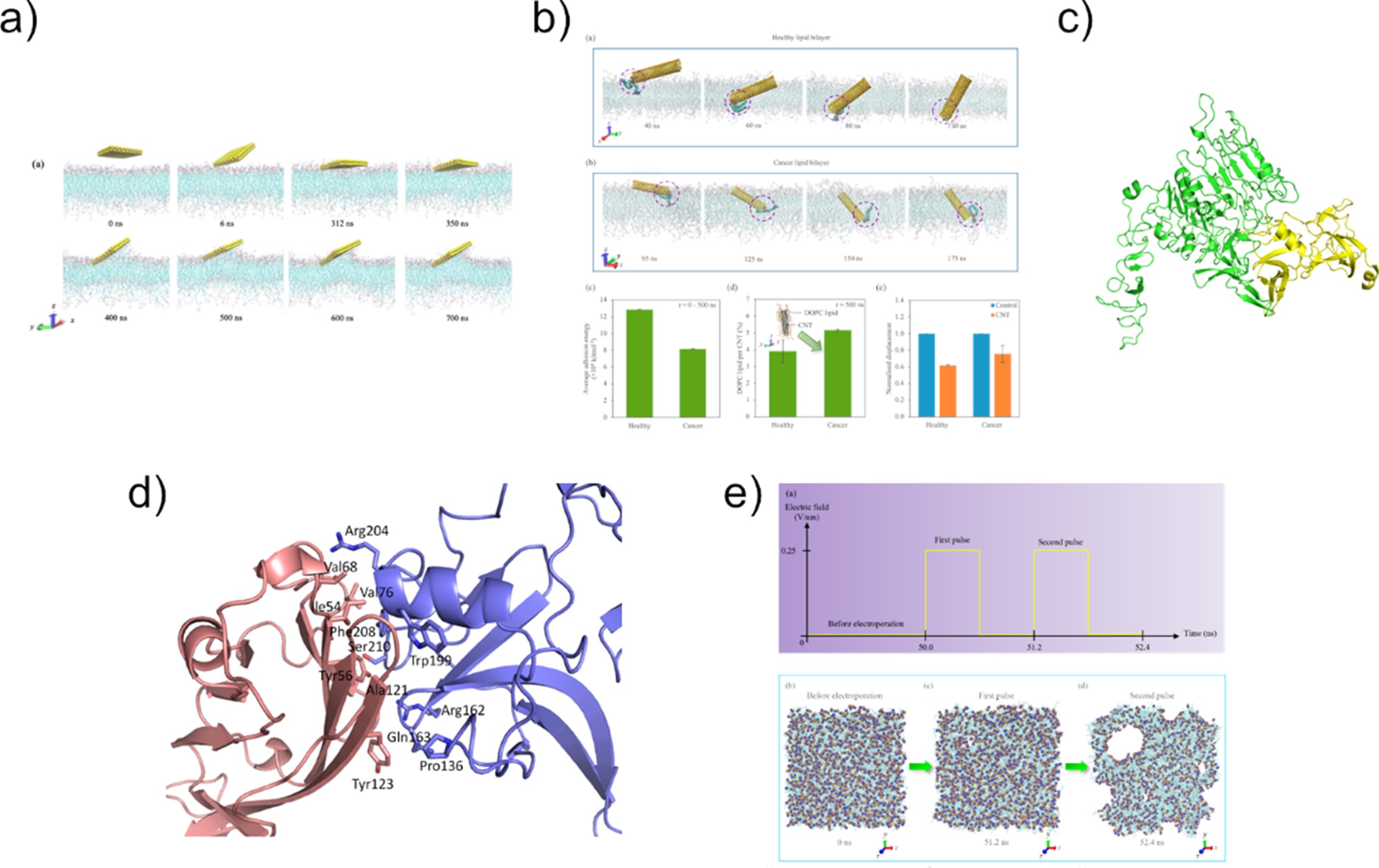
Figure 3.Bioelectronics for biomedical applications. (a) MoS2 nanosheet-containing DC resistance sensor for detection of cancer cells (Nanoscale Adv 2021). (b) Carbon nanotube-containing photo-assisted AC pulse sensor for detection of cancer cells (ACS Omega 2022). (c) WS2-PEG-phage bioelectronic sensor for breast cancer cell detection (Nanoscale 2023). (d) Electrothermal therapy for pancreatic cancer mediated by M13 bacteriophage (Pharmaceutics 2023). (e) Effect of electric pulses on membrane permeabilisation (Nanoscale 2022).
| Principal Scientist | TAN Yaw Sing | [View Bio] |
| Scientist | NG Tze Yang Justin |
| Scientist | MODEE Rohit Laxman |
| Research Officer (T-UP with QDX) | CHONG Kian Chee |
| Research Officer | TAN Zi Ting Eunice |
 | TAN Yaw Sing Principal Investigator Email: tan_yaw_sing@a-star.edu.sg Research Group: Computational Chemical Biology and Fragment-Based Design |
Dr. Tan Yaw Sing graduated with a B.Sc. (Hons) from the National University of Singapore in 2009, with a double major in Chemistry and Life Sciences. Funded by the A*STAR Graduate Scholarship, he proceeded to pursue his postgraduate studies at the University of Cambridge, where he obtained his PhD in Chemistry in 2014. In the same year, he joined Dr. Chandra Verma’s group at the Bioinformatics Institute (BII) as a postdoctoral research fellow. He has been leading his own group in BII since April 2021 and has been appointed as Principal Investigator in April 2024.
Research Interests
Dr. Tan’s research interests span the diverse fields of computational chemical biology, computer-aided drug design, and computational structural biology. He hopes to transform current strategies for drug discovery and chemical biology by improving the accuracy of binding site prediction, providing new insights into the structure and dynamics of drug targets, and guiding the rational design of therapeutics and chemical tools.
Group Members
| Scientist | NG Tze Yang Justin |
| Scientist | MODEE Rohit Laxman |
| Research Officer (T-UP with QDX) | CHONG Kian Chee |
| Research Officer | TAN Zi Ting Eunice |