
Working at the interface between physics and biology, we develop physical models and computational approaches for modelling structures of biomolecules and for exploring their functional characteristics. The research topics in the group include, but are not limited to, protein allostery, biophysics of chromatin and epigenetic regulation, evolution of protein function, evolutionary-inspired design of protein function, and molecular mechanisms of adaptation
Applying our 3D whole-genome chromatin ensemble reconstruction [1], we tackled genomic mechanisms of anosmia, a potential diagnostic indicator for early-stage COVID-19 infection onset. Obtaining the chromatin ensemble reconstruction and analysing the fine structural hierarchy of chromatin in olfactory sensory neurons, we showed how structural modifications on different levels of chromatin organization in COVID-19 patients can be associated with the pathology [2]. We proposed further steps for developing highly sensitive diagnostics and efficient treatments in genomic and epigenetic studies of infectious diseases with long-term ramifications.
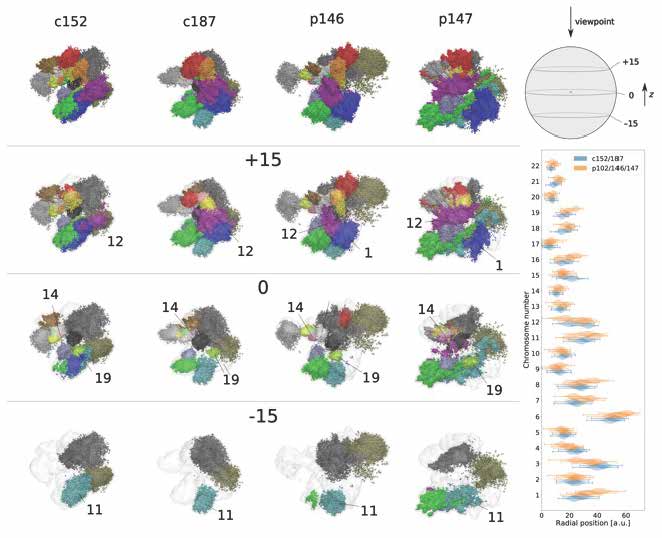
Figure 1. Reconstructions of whole-genome chromatin ensemble for olfactory sensory neurons in SARS-CoV-2-infected patients and controls.
Do we need to talk about evolutionary-inspired protein design in times of AlphaFold? We argue that it is still important, taking a “back in time travel” and showing how simple elementary functions arrived from the prebiotic world became a cornerstone of multiple proteins [3]. Focusing on phosphate binding, one can show how its reconstructed ancient prototype can give rise to all relevant phosphate-binding signatures in different functions, revealing versatility towards evolutionary design and functional diversification. Respectively, the whole repertoire of evolutionary selected and weathered diversity of elementary functions, from which the whole diversity of contemporary biological functions can be obtained [4], should be used together with and complemented by AlphaFold predictions to facilitate rational design of desirable protein functions.
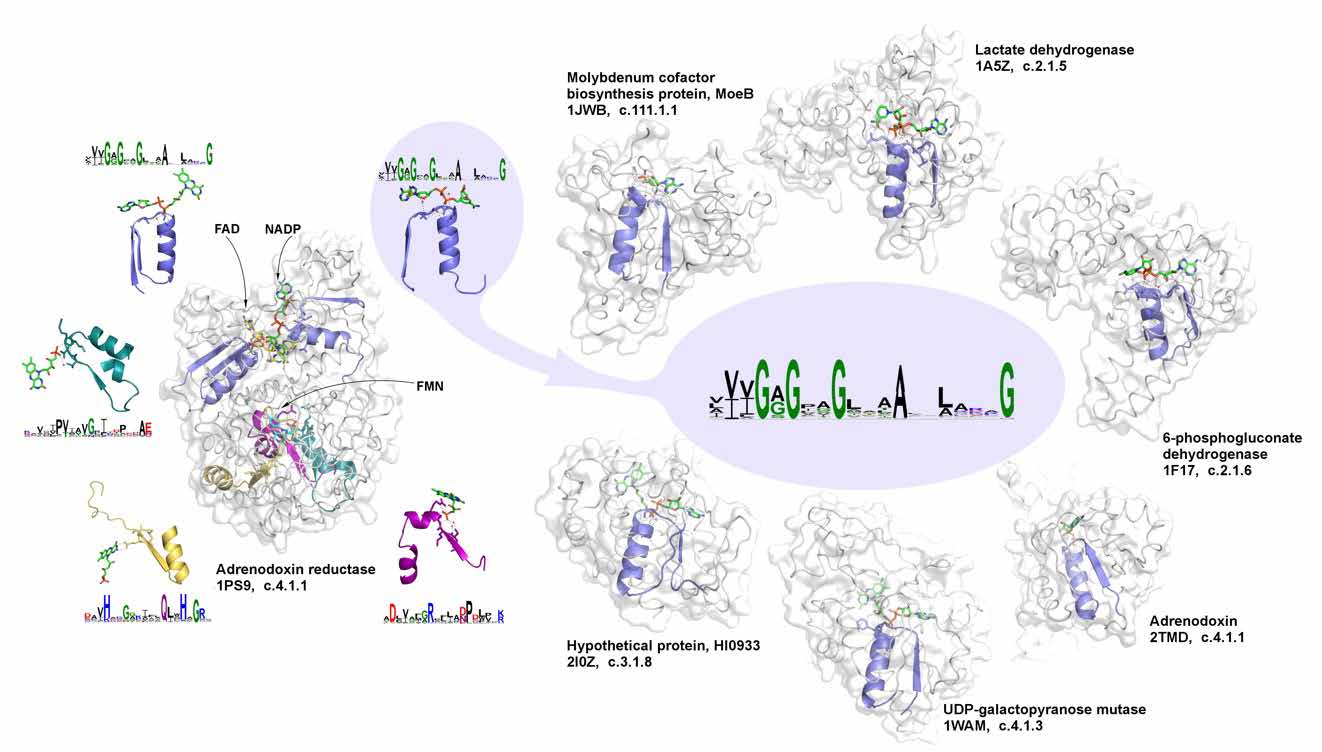
Figure 2. The signature of phosphate binding in dinucleotide-containing ligands and its representatives in different biochemical functions.
Thinking of engineering and design of stable and functional proteins, one should always “ask” Nature about recipes to achieve it. Using a wealth of currently available complete genomes/proteomes, we explored signatures of adaptation to three environmental factors, pH, salinity, and temperature. Our work also revealed an evolutionary perspective on the adaptation mechanisms by means of the comparative analysis of genomic/proteomic traits of archaeal and bacterial species [5]. Showing major trends in compositions of proteins and nucleic acids in response to harsh environments, we point to corresponding mechanisms of stability, their specifics depending on the environmental factors and their combinations, and the influence of the evolutionary history of the host organisms. We also formulate outstanding questions, which will be important to address in engineering proteins with desirable functions and regulatory mechanisms in individual and combined extreme environments encountered in different biotechnological applications.
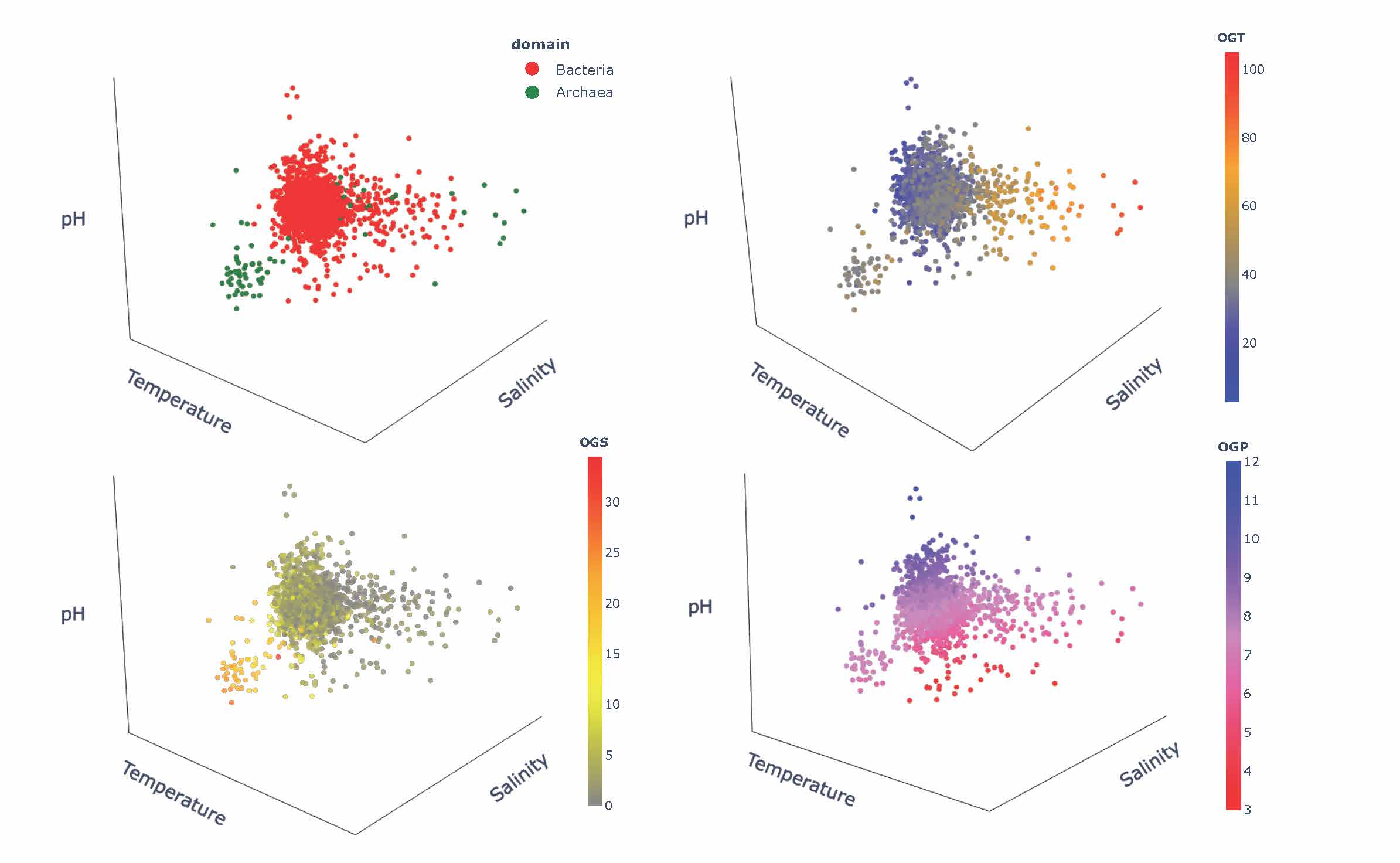
Figure 3. The 3D Principal Component Analysis-based illustration of the complexity of adaptation to extreme environments and their combinations.
Continuing our work on allostery [6-11], we proposed a sequence-dependent version [12] of the structure-based statistical mechanical model of allostery (SBSMMA) developed earlier in the group. It includes the Miyazawa-Jernigan residue-residue potential allowing to operate with amino acid residue types and to explore allosteric effects of their substitutions. Working towards important biomedical applications of allostery [13] – design of allosteric drugs, we describe important characteristics of allosteric sites, effectors, and their modes of actions distinguishing them from the orthosteric counterparts thus calling for new principles and protocols in the quests for allosteric drugs [14]. Showing the importance of considering both binding affinity and allosteric signalling in establishing the structure-activity relationships (SARs) toward design of allosteric effectors, we argued that pairs of allosteric sites and their effector ligands – the site-effector pairs - should be generated and adjusted simultaneously in the framework of what we call directed design protocol. We also discussed key ideas and approaches for designing allosteric effectors including reverse perturbation, targeted and agnostic analysis.
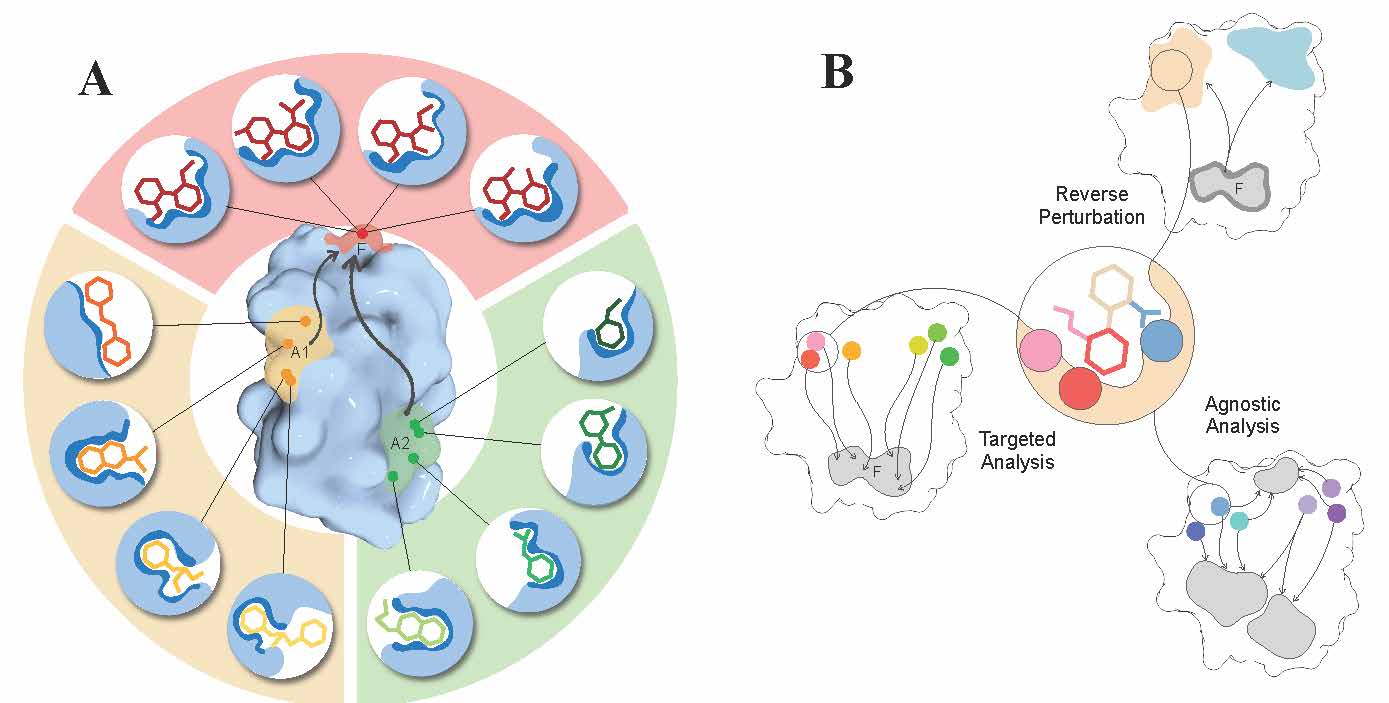
Figure 4. Allosteric versus orthosteric drugs and key ideas for their design.
A. Allosteric versus orthosteric sites, ligands, and modes of actions. Orthosteric ligands exert their effects by binding to the same active/functional site (red, F). Therefore, the ligands and their interactions with residues in the functional site typically exhibit a high level of conservatism. On the other hand, allosteric effectors bind non-competitively to remote allosteric sites (A1, A2 in orange and green). There can be multiple (non-)overlapping sites within locations (A1, A2) that are allosterically linked to the regulated F site, providing a diverse array of site-effector pairs that can differently modulate the function. Based on desired allosteric modulation, one can screen/design and further fine-tune site-effector pairs to originate specific allosteric signalling. B. The reverse perturbation, targeted, and agnostic analysis – a foundation for fragment-based design of allosteric effectors. The reverse perturbation technique allows to detect locations (beige and light blue) that are allosterically connected with the functional site (F) upon simulating substrate binding at the latter. The targeted analysis identifies and shortlists residues that originate the optimal/desired allosteric signalling to targeted residues in F, as a result of perturbations such as mutations or small-probe binding. The agnostic analysis reveals residues that are extensively coupled to at least one location in protein by exploring and mapping all allosteric connections in the protein.
| Senior Principal Investigator | BEREZOVSKY Igor N. | [View Bio] |
| Senior Scientist | SU Tran-to Chinh |
| Scientist | TEE Wei Ven |
| Scientist | DONG Bingxue |
| Research Officer | RAECHELL |
 | BEREZOVSKY Igor N. Senior Principal Investigator Email: igorb@a-star.edu.sg Research Group: Physics and Evolution of Biological Macromolecules |
Igor Berezovsky studied physics at the Moscow Engineering Physics Institute (MSc, 1993) and obtained PhD in physics and mathematics from the Moscow Institute of Physics and Technology (1997). He started his scientific career at the Engelhardt Institute of Molecular Biology (Moscow) where he conducted his MSc and PhD research, then worked as a research fellow (until 1998). After postdoctoral researcher at the Weizmann Institute of Science (1999-2002) and the Harvard University (2003-2006), Igor was a senior scientist/group leader at the Bergen Center for Computational Science, University of Bergen (Norway) before joining the Bioinformatics Institute in January 2014.
| Senior Scientist | SU Tran-to Chinh |
| Scientist | TEE Wei Ven |
| Scientist | DONG Bingxue |
| Research Officer | RAECHELL |